
Im Mai 2016 wurde die deutsche Ausgabe der IEC 60601-1-2:2014 (Edition 4) als DIN EN 60601-1-2:2016 mit dem Titel Elektromagnetische Störgrößen – Anforderungen und Prüfungen veröffentlicht.
Ende 2020 folgte die um das Amendment 1 (AMD 1) ergänzte und als Edition 4.1 bezeichnete Ausgabe dieser „EMV-Norm“.
Medizinproduktehersteller, die in den Anwendungsbereich der IEC 60601-1-2 fallen, sollten die Änderungen zwischen den beiden Editionen der Norm kennen. Denn sie müssen entscheiden, ob neue EMV-Prüfungen notwendig sind oder sogar ein Re-Design der Produkte, um die Konformität ihrer Produkte zu gewährleisten.
Das Entscheidungsdiagramm im Kapitel 5. („Tipps“) hilft dabei.
1. Um was es geht: Elektromagnetische Verträglichkeit
a) Elektromagnetische Störfestigkeit
Medizinprodukte müssen sicher sein. Sie dürfen beispielsweise nicht ausfallen oder Fehlfunktionen aufweisen, nur weil ein anwendender Arzt telefoniert und die Handy-Strahlung das Medizinprodukt stört. Daher verlangt die IEC 60601-1-2 die „elektromagnetische Störfestigkeit“ von Medizinprodukten.
b) Elektromagnetische Aussendungen
Umgekehrt darf es auch nicht sein, dass Medizinprodukte durch „elektromagnetische Aussendungen“ die Umgebung negativ beeinträchtigen und Störwirkung haben auf andere Medizinprodukte, Radios, das Telefon- und Funknetz, Computer oder die Stromversorgung.
c) Störfestigkeit + Aussendungen = Verträglichkeit
Die IEC 60601-1-2 bestimmt die Anforderungen an die elektromagnetische Verträglichkeit (EMV), weshalb man oft von der EMV-Norm spricht.
Die Norm definiert den Begriff:
„Fähigkeit eines „ME-Gerätes“ oder „ME Systems“, in seiner elektromagnetischen Umgebung zufriedenstellend zu funktionieren, ohne in diese Umgebung, zu der auch andere Einrichtungen gehören, unzulässige elektromagnetische Störgrößen einzubringen.“
Quelle: IEC 60601-1-2
Somit ist die Norm auch geeignet, die MDR-Anforderungen an Interoperabilität und Kompatibilität bezüglich der elektromagnetischen Umgebung zu erfüllen; sie gilt als harmonisierte Norm.
2.) Die IEC 60601-1-2 im Überblick
a) Geltungsbereich
Medizinisch-elektrische Geräte
Die IEC 60601-1-2 stellt den Stand der Technik dar für alle Hersteller, deren Medizinprodukte oder deren Zubehör unter den Anwendungsbereich der Basisnorm IEC 60601-1 fallen. Das sind die medizinisch-elektrischen Geräte und Systeme, kurz ME-Geräte und ME-Systeme.
Die Norm ist also anzuwenden (insbesondere) auf Medizinprodukte sowie Zubehör, wenn diese über ein Anwendungsteil verfügen. Das ist der Teil des Medizinprodukts, der bei bestimmungsgemäßem Gebrauch in physischen Kontakt mit dem Patienten kommen muss.
Die Anwendbarkeit ist aber nicht auf ME-Geräte und ME-Systeme beschränkt. Es gibt auch elektrische Geräte für die Nutzung in einer medizinischen Umgebung, die bestimmungsgemäß kein Medizinprodukt sind; z. B. einen Visiten-PC auf der Intensivstation oder einen Wandmonitor in einem OP. Auch deren Hersteller sollten die IEC 60601-1-2 erfüllen und damit sicherstellen, dass andere Medizinprodukte nicht beeinflusst werden. Häufig werden diese Nicht-Medizinprodukte unter der Niederspannungsrichtlinie in Verkehr gebracht und erhalten in der Konformitätserklärung den Zusatz „medical grade“.
Dieser Artikel verschafft Ihnen eine schnelle Übersicht über die Anwendbarkeit und die Anforderungen der IEC 60601-1.
Implantate
Die IEC 60601-1-2 ist nicht für Implantate nicht anwendbar, aber für deren Zubehör, dass ein Implantat von außen überwacht oder steuert. (Implantate haben eigene Normen Bsp. ISO 14117.)
IVD-Geräte, die ebenfalls Medizinprodukte sind
Für IVD-Produkte, die in einem Labor betrieben werden, gelten ebenfalls Anforderungen an die Störfestigkeit und Störausstrahlung. Die dafür zuständige Norm ist aber nicht die IEC 60601-1-2, sondern die Norm IEC 61326-2-6 für Laborequipment. IVD-Produkte, die im medizinischen Umfeld betrieben werden (sogenannte Point of Care IVD), sollten sich aber an den Prüfschärfegraden der IEC 60601-1-2 orientieren.
Beide Normen, die IEC 60601-1-2 und die IEC 61326-2-6, greifen bei der Spezifikation der Prüfungen auf den gleichen Satz an Fachgrundnormen zu: Das sind die IEC-61000-3-Grundnormen für Emission und die IEC-61000-4-Grundnormen für Immunität. Die IEC 60601-1-2 und die IEC 61326-2-6 spezifizieren für diese Prüfung ihrerseits spezifische Grenzwerte oder Prüfpegel.
Medizinprodukte, die auch Funktechniken verwenden
Diese Produkte müssen neben den Anforderungen der MDR auch die Anforderungen der Richtlinie RED (Radio Equipment Directive) erfüllen. Für Produkte mit Funktechnik ist die Anwendung der IEC 60601-1-2 allein nicht ausreichend, um die Anforderungen der RED zu erfüllen. Daher gelten weitere Normen, die hauptsächlich unter der Schirmherrschaft der europäischen Kommission ETSI (Europäisches Institut für Telekommunikationsnormen) geschrieben werden und kostenlos zur Verfügung stehen. Spannend ist, dass diese Normen neben der elektromagnetischen Koexistenz auch die biologische Auswirkung der Strahlung (biologische Gefährdungen) berücksichtigen, was die IEC 60601-1-2 völlig ignoriert.
Medizinprodukte, die in speziellen Umgebungen betrieben werden
Für Medizinprodukte, die in der Flugrettung oder in Krankenfahrzeugen betrieben werden, gelten erweiterte Anforderungen: zum Beispiel die RTCA DO-160 oder der ISO 14708 für Implantate in einer MRI-Umgebung.
b) Anforderungen der EMV-Norm
Die IEC 60601-1-2 legt fest,
- mit welchen Arten von elektromagnetischen Störungen Medizinprodukte problemlos umgehen müssen,
- wie stark Medizinprodukte selbst elektromagnetische Strahlung aussenden dürfen,
- wie die diesbezüglichen Anforderungen von der spezifizierten Nutzungsumgebung und vom Risiko des Produkts abhängig sein dürfen,
- wie Hersteller bzw. Prüflabore dies messen, prüfen und diese Messungen dokumentieren müssen und
- wie die Hersteller das Produkt kennzeichnen und welche Informationen in der Gebrauchsanweisung stehen müssen.
c) Aufbau der IEC 60601-1-2
Gemäß diesen Forderungen ist die Norm aufgebaut:
- Kapitel 6 legt die Dokumentation der Prüfungen fest.
- Kapitel 7 stellt Anforderung an die „Aussendung“ (Emission) von elektromagnetischer Strahlung.
- Kapitel 8 bestimmt die Anforderungen an die Störfestigkeit (Immission).
- Kapitel 9 nennt die Anforderungen an Prüfberichte.
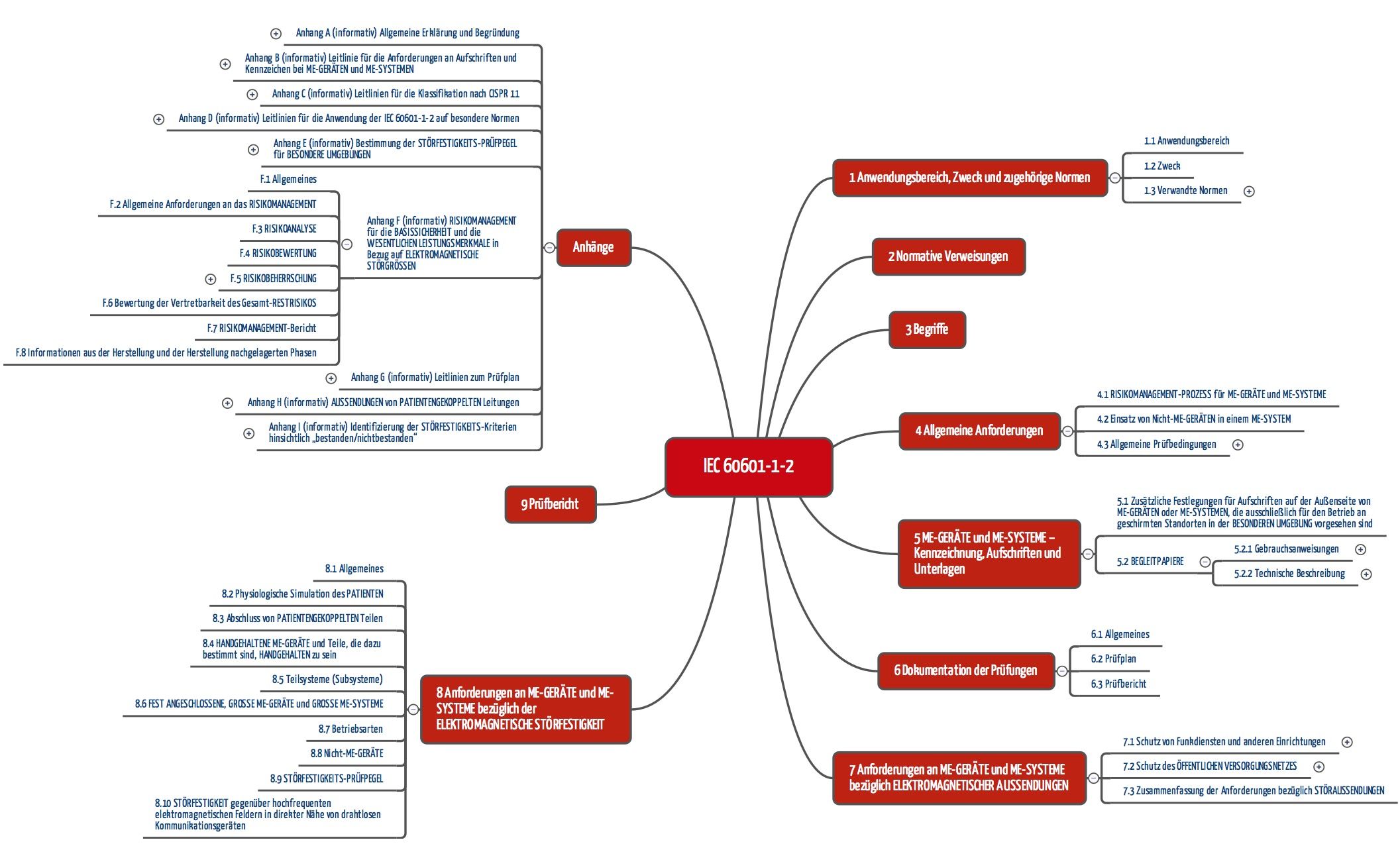
Besondere Beachtung verdient der Anhang F, der die enge Verzahnung der Norm mit dem Risikomanagement (nach ISO 14971) offenbart. Dieser Fokus auf das Risikomanagement ist neu seit der 4. Edition und wird mit der Version 4.1 noch besser erläutert.
3. Unterschiede zwischen den Editionen der Norm
a) Unterschiede zwischen der 3. und 4. Edition der IEC 60601-1-2
Die vierte Edition der IEC 60601-1-2 unterscheidet sich deutlich von der Vorgängerversion. Das offenbart bereits ein Blick ins Inhaltsverzeichnis.
Zu den Änderungen zählen:
- Anwendung der Risikoanalyse als Begründung für das Festlegen von Testlevels
- EMV als integraler Bestandteil in der Risikoanalyse (Einfluss auf Basissicherheit und wesentliche Leistungsmerkmale)
- Testlevels für Immunität sind abhängig von der Einsatzumgebung:
- Professional Healthcare (Praxen, Kliniken, …)
- Home Healthcare (Wohnungen, Geschäfte, öffentliche Wege und Gebäude, Fahrzeuge)
- Special (MRT, Militär, Schwerindustrie)
Die Änderungen haben einen wesentlichen Einfluss auf das Design und die Architektur von Medizinprodukten. Vergleichbar mit den Änderungen bei der Einführung der IEC 60601-1 mit der dritten Edition hat die Risikoanalyse auch hier einen wesentlichen Stellenwert in der Norm erhalten.
Hersteller müssen die wesentlichen Leistungsmerkmale ihrer Geräte präzise bestimmen, um zuverlässige Performanzkriterien im EMV-Testplan festzulegen zu können.
b) Unterschiede zwischen den Editionen 4.0 und 4.1
Das AMD 1 vom Jahresende 2020 ergänzt die 4. Version der Norm. Zusammen bilden beide die Version 4.1.

Motivation für und Überblick über die Änderungen
Das Normenkomitee hat insgesamt 15 Änderungen identifiziert, die es für dringlich hält und die nicht bis zur fünften Ausgabe der Norm warten konnten. Die wichtigsten Änderungen sind:
- Neue digitale Technologien (RF-Transmitter)
Die Menge und Dichte von RF-Transmittern (z. B. WiFi, Bluetooth-Verbindungen, DECT, RFID, LTE, 5G, Wireless Charging etc.) hat sich massiv erhöht. Neue Antennentopologien und höhere Übertragungsraten haben zu einem komplexeren und vielfältigeren Nutzungs- und Belastungsmuster geführt. Ebenso hat die vermehrte Tragbarkeit von Geräten den Abstand zwischen den Quellen der abgestrahlten HF-Energie und den Geräten, die durch diese Energie gestört werden könnten, drastisch verringert.
Das hat die Autoren dazu veranlasst, die Anforderungen an die Störfestigkeit für Geräte, die in unmittelbarer Nähe von Störquellen verwendet werden, zu erhöhen. Beispiele sind Insulin- oder Schmerzpumpen, die am Körper getragen werden und deren Ausfall nicht vertretbar wäre. - Verständlichkeit der Edition 4.0
Der in der vierten Ausgabe deutlich überarbeitete Anhang F hat offensichtlich einige Fragen aufgeworfen. Daher hat das Autoren-Team die Verständlichkeit dieses Anhangs erhöht. Jede Referenz an das Risikomanagement wurde kommentiert und mit Beispielen ergänzt. - Schwierigkeiten bei der Anwendbarkeit bei großen Geräten
Großgeräte wie Kernspingeräte lassen sich nicht einfach in einem EMV-Labor testen. Daher gibt die Edition 4.1 spezifische Hinweise für die Prüfung dieser Medizinprodukte. - Veraltete Verweise
Die IEC 60601-1-2 arbeitet mit datierten Verweisen. Da inzwischen Normen wie die ISO 14971 in einer neuen dritten Ausgabe vorliegen, mussten diese Verweise aktualisiert werden.
Neue Prüfung nach IEC 61000-4-39
Für die Prüfung der Störfestigkeit im Nahfeld von typischen HF-Transmittern wie Mobiltelefonen, Bluetooth oder WLAN waren bisher die Prüfmethoden der Norm IEC 61000-4-3 –Testing and measurement techniques – Radiated, radio-frequency, electromagnetic field immunity test – maßgebend.
Die Autoren der IEC 60601-1-2 haben aber schon beim Schreiben der vierten Edition in der Begründung zum Abschnitt 8.10 darauf hingewiesen, dass die Prüfung nach IEC 61000-4-3 allein nicht mehr ausreichend ist. Sie haben begonnen, ein weiteres Prüfverfahren zu erarbeiten, welches aber zum Zeitpunkt der Veröffentlichung der 4. Edition noch nicht validiert war.
Diese Prüfung gilt speziell für Geräte oder Systeme, die in unmittelbarer Nähe (< 15 cm) Störquellen ausgesetzt sind. Damit soll das Risiko durch störempfindliche Teile wie Spulen, Signaltransformatoren und Halleffekt-Sensoren weiter gesenkt werden. Die IEC 61000-4-39 ergänzt daher die IEC 61000-4-3 und ersetzt diese nicht.
Bei der Prüfung werden Störfelder, die für typische Applikationen im Nahfeld charakteristisch sind, simuliert. Dazu zählen:
- Induktionskochgeräte und Backöfen in der häuslichen Umgebung
- RFID-Transponder und Lesegeräte
- z. B. Chirurgie zur Identifikation von Instrumenten
- z. B. Chirurgie zur Vermeidung, dass Tupfer oder Schwämme im Patienten verbleiben
- Geräte zur Lageerkennung (z. B. in Katheterlabors)
- Systeme zur drahtlosen Energieübertragung für Elektrofahrzeuge
Im AMD 1 werden diese neuen Prüfverfahren im Abschnitt 8.11 (IMMUNITY to proximity magnetic fields in the frequency range 9 kHz to 13,56 MHz) eingeführt.
Damit wird klar, dass die Autoren die Norm schrittweise an die sich ändernde Realität anpassen, um weiterhin den Stand der Technik zu repräsentieren.
Prüfen von großen Systemen
Die IEC 60601-1-2 bietet bisher drei Methoden zur Prüfung der Störfestigkeit und Störausstrahlung von großen und fest installierten ME-Geräten bzw. ME-Systemen an. Dabei definiert die EMV-Norm „groß“ als größer als 2 m × 2 m × 2,5 m (z. B. Anlagen zur Turmorbestrahlung oder MRIs).
Für Geräte dieser Größenordnung bietet die Norm diese Optionen an:
- Als Gesamtsystem im Testhaus
- Als Teilsysteme im Testhaus
- Beim Betreiber vor Ort (in situ) nach der Installation und vor der Inbetriebnahme
Die Autoren räumen ein, dass das Testen von Teilsystemen oder Testen beim Betreiber vor Ort oft ungeeignet oder nicht möglich ist. Manche Länder lassen vielleicht auch nur Importe von Geräten bzw. Systemen zu, die bereits eine gültige EMV-Prüfung nachweisen können.
Das AMD 1 ermöglicht daher nun auch die Prüfung in den Räumlichkeiten des Herstellers. Bei den Herstellern sind die Zusatzeinrichtungen, die zur Steuerung und Überwachung des Prüflings notwendig sind, in der Regel verfügbar.
Dadurch ist die Prüfung in einer repräsentativen Konfiguration möglich, die ebenfalls die Anforderung zur Prüfung im Betriebsmodus erfüllt. Die Prüfung beim Hersteller kann somit als gleichwertig mit der Prüfung vor Ort angesehen werden.
Bei der Prüfung beim Hersteller muss die gute Praxis für die EMV-Messung eingehalten werden. Wenn die anwendbaren EMV-Grundnormen eine Prüfung an Ort und Stelle erlauben, haben die Anforderungen in den EMV-Grundnormen Vorrang.
Zusätzlich sollte eine Begründung, warum die Prüfung der ME-Geräte bzw. ME-Systeme vor Ort beim Hersteller gerechtfertigt ist, im Prüfplan angegeben und im Prüfbericht dokumentiert werden.
Hersteller können EMV-Prüfhäuser beauftragen, wenn diese technisch und fachlich dafür eingerichtet sind, beim Hersteller vor Ort zu prüfen.
Zusammenspiel mit dem Risikomanagement
Der neue Anhang F enthält eine Tabelle, die sich über sechs Seiten erstreckt. Sie liefert für alle Abschnitte der Norm, die Anforderungen an das Risikomanagement enthalten, eine Begründung zu den Überlegungen. Es wird auch beschrieben, in welcher Reihenfolge die Risikomanagement-Aktivitäten durchgeführt werden sollten.
Das ist zu begrüßen, da viele Hersteller Mühe hatten, das Risikomanagement im Kontext der EMV umzusetzen. Auch die Prüfhäuser taten sich mit der Kontrolle der risikominimierenden Maßnahmen schwer.
Sie sollten in der Lage sein zu erklären, wie von den angewendeten oder angenommenen Risikominderungen vernünftigerweise erwartet werden kann, dass sie über die spezifizierte Betriebslebensdauer und in allen spezifizierten Nutzungsumgebungen wirksam bleiben.
Wenn Sie zum Beispiel die ESD-Prüfpegel reduzieren, weil Ihr Gerät an einem Ort mit kontrollierter Luftfeuchtigkeit betrieben wird, müssten Sie diskutieren, ob das Gerät auch außerhalb dieser Umgebung verwendet werden könnte. Auch Bauteile wie Filterkondensatoren altern schneller, wenn sie höheren Temperaturen oder einem anderem physikalischen Stress ausgesetzt sind.
Bei der Risikoanalyse geht es nicht immer um das Abschätzen von Wahrscheinlichkeiten. Auch einfache Ja-Nein-Überlegungen gehören zur Risikoanalyse: z. B. ob eine Gefährdungssituation oder die Verschlechterung oder der Ausfall eines Bauteils wahrscheinlich genug ist, um betrachtet zu werden. Genauso ist die Festlegung der EMV-Umgebung eine Aktivität der Risikoanalyse. Es müssen also nicht alle Fragestellungen in der Risikotabelle dokumentiert werden.
4. Übergangsfristen in der EU und den USA
a) Europa
Übergang von der Edition 3 auf die Edition 4
Die Harmonisierung wurde für Ende 2016/Anfang 2017 erwartet. Die Tatsache, dass der Beuth-Verlag die DIN EN 60601-1-2 bereits veröffentlich hat, bestärkt diese Vermutung.
Da der Harmonisierungsprozess zum Erliegen gekommen ist, sollten Medizinproduktehersteller bei einer Entwicklung in jedem Fall auf die 4. Edition wechseln. Eine spätere Änderung des Designs wäre zu aufwendig.
Die Benannten Stellen fordern diese Version der Norm mit Verweis auf den Stand der Technik ein. Falls es zu einer Harmonisierung der Edition 4 kommt, müssen neue Geräte die Forderungen der vierten Edition der IEC 60601-1-2 erfüllen – wahrscheinlich ohne weitere Übergangsfrist.
Für bereits im Markt befindliche Geräte sowie Geräte, die weiterhin unverändert produziert werden, akzeptieren die Benannten Stellen bestehende EMV-Prüfungen. Sie verlangen aber eine kontinuierliche Aktualisierung der Risikomanagementakte, die auch Risiken aufgrund von elektromagnetischen Störungen bewerten müssen.
Beachten Sie, dass sich neben der IEC 60601-1-2 weitere Normen geändert haben (wie die IEC 60601-1 Version 3.1) oder ändern werden.
Sie finden hier eine Übersicht über geplante Änderungen an der Normenfamilie IEC 60601.
Übergang von der Edition 4 auf die Edition 4.1
Die EU hat kürzlich den Entwurf Standardisation requests to the European standardisation organisations erstellt und darin für alle MDR- und IVDR-Produkte diejenigen Normen aufgelistet, die die Normenkommissionen bis 27. Mai 2024 harmonisieren müssen (Z-Anhänge schreiben). Darin befindet sich aber erst die 4. Edition der EMV-Norm.

Da unter der MDR noch keine Norm harmonisiert ist, müssten Hersteller wie bereits dargelegt den Stand der Technik berücksichtigen. Da das Amendment bei der FDA mit Einschränkungen bereits gelistet ist, dürfte dies genau diesen Stand reflektieren.
Das Johner Institut empfiehlt daher, die Edition 4.1 der IEC 60601-1-2 anzuwenden. Zudem entspricht die Norm auch eher der Realität und macht somit Medizinprodukte zuverlässiger.
(Auch wenn manche Experten behaupten, dass die Norm immer noch realitätsfremd sei. Dessen sind sich wahrscheinlich auch die Autoren bewusst, nur darf die Verschärfung der Norm nicht dazu führen, dass ein Versorgungsengpass bei Medizinprodukten entsteht.)
b) USA
Übergang von der Edition 3 auf die Edition 4
Die FDA akzeptiert die 4. Edition der IEC 60601-1-2 mit kleinen Ausnahmen bereits seit 2014. Nach dem 31.12.2018 wurde die Anwendung der 4. Edition der EMV-Norm verbindlich.
Im Guidance-Dokument Design Considerations for Devices Intended for Home Use wird die Verwendung der vierten Edition für Geräte in häuslicher Umgebung bereits empfohlen (zumindest die Anwendung der Testkriterien).
Die Anwendung der Version 3.1 der IEC 60601-1 wurde schon früher, verbindlich nämlich am 1. August 2016.
Übergang von der Edition 4 auf die Edition 4.1
Die FDA führt das AMD 1 bereits in der Liste der Recognized Standards. Nach dem 17. Dezember 2023 werden andere Konformitätserklärungen nicht mehr akzeptiert.
Die FDA hat im Juni 2022 ihr 20-seitiges Guidance-Dokument Electromagnetic Compatibility (EMC) of Medical Devices veröffentlicht. Darin wird bereits das Prüfen von RFID-Systemen nach AIM 7351731 gefordert.
Bei Hunderten an Normen, Gesetzen, Verordnungen, Richtlinien und Leitlinien ist es schwer, den Überblick über Neuerungen und Änderungen zu behalten.
Nutzen Sie das Regulatory Radar des Johner Instituts, das Ihnen diese ständige Recherche abnimmt und Sie bei Änderungen proaktiv informiert. Das erspart Ihnen nicht nur Arbeit und Kosten; es hilft Ihnen, die gesetzlichen Anforderungen zu erfüllen und regulatorischen Ärger zu vermeiden.
5. Tipps zur Vermeidung von regulatorischem Ärger
Diese Tipps beinhalten allgemeine Hinweise, die Sie im Rahmen der PMS-Aktivitäten bei jeder Änderung der Norm immer berücksichtigen sollten. Weiterhin gibt es Hinweise, die sich auf den Wechsel zur Edition 4.1 beziehen.
Tipp 1: Dokumentation auf Stand bringen
Um einen EMV-gerechten Entwurfsprozess nachzuweisen, sollten Sie Ihre Überlegungen in einem EMV-Konzept dokumentieren. Dazu gehören z. B. die Analyse der Umgebung und die Identifikation von typischen Störquellen in der spezifizierten Umgebung, andere Medizinprodukte eingeschlossen. Darin sollte das Risiko bewertet werden, das von jeder möglichen Fehlfunktion oder Unterbrechung ausgehen kann. Ein Beispiel für ein EMV-Konzept finden Sie in unserem Praxisleitfaden zur IEC 60601-1. Weitere Anleitungen finden Sie in dem Leitfaden Electromagnetic Compatibility for Functional Safety.
Sie sollten ggf. die Zweckbestimmung überprüfen oder ergänzen, beispielsweise indem Sie die Nutzungsumgebung und die Nutzung präziser spezifizieren (z. B. tragbar oder am Körper getragen).
Überprüfen Sie, ob die Hinweise in der Gebrauchsanleitung gemäß den neuen Vorgaben der EMV-Norm sind.
Tipp 2: Risikomanagement betreiben
Führen Sie eine Gap-Analyse Ihrer bestehenden Risikomanagementakte gegen die geänderten und neuen Anforderungen der aktuellen Versionen der Normen durch. Ein Ergebnis der Überarbeitung der Risikoanalyse sollte sein,
- ob Sie nichts machen,
- die Prüfung nur wiederholen, um die Konformität zu demonstrieren, oder
- eine Änderung am Gerät durchführen und ebenfalls die Prüfung wiederholen oder
- Ihre Technische Dokumentation wie Risikoanalyse, Gebrauchsanweisung, Labeling oder Klinische Bewertung aktualisieren müssen.
Dokumentieren Sie die Ergebnisse im PMS-Bericht und ggf. auch im PSUR-Bericht.
Der Anhang F enthält mit der Grafik F.1 eine Empfehlung für die Reihenfolge der Aktivitäten im Risikomanagement. Die Aktivitäten könnten Sie auch gleich in Ihren Entwicklungsplan einbauen und so zeigen, dass Sie einen EMV-gerechten Entwurfsprozess einhalten.
Tipp 3: Vorgehen mit Benannter Stelle abstimmen
Wenn Sie bestehende Geräte weiterhin unverändert produzieren, sollten Sie die Ergebnisse aus der Risikoanalyse und Ihr geplantes Vorgehen mit Ihrer Benannten Stelle (notified body) besprechen. Klären Sie ab, welche Maßnahmen von Ihnen erwartet werden, wenn Sie beispielsweise kleine Änderungen an Produkten vornehmen.
Tipp 4: Elektromagnetische Kompatibilität prüfen und Gaps identifizieren
Führen Sie EMV-Vortests mit bestehenden Geräten durch um festzustellen, ob das aktuelle Design die geänderten Anforderungen erfüllt. Das Labor, bei dem Sie diesen Vortest durchführen, muss nicht notwendigerweise akkreditiert sein.
Tipp 5: Prüfen, welche Geräte betroffen sind
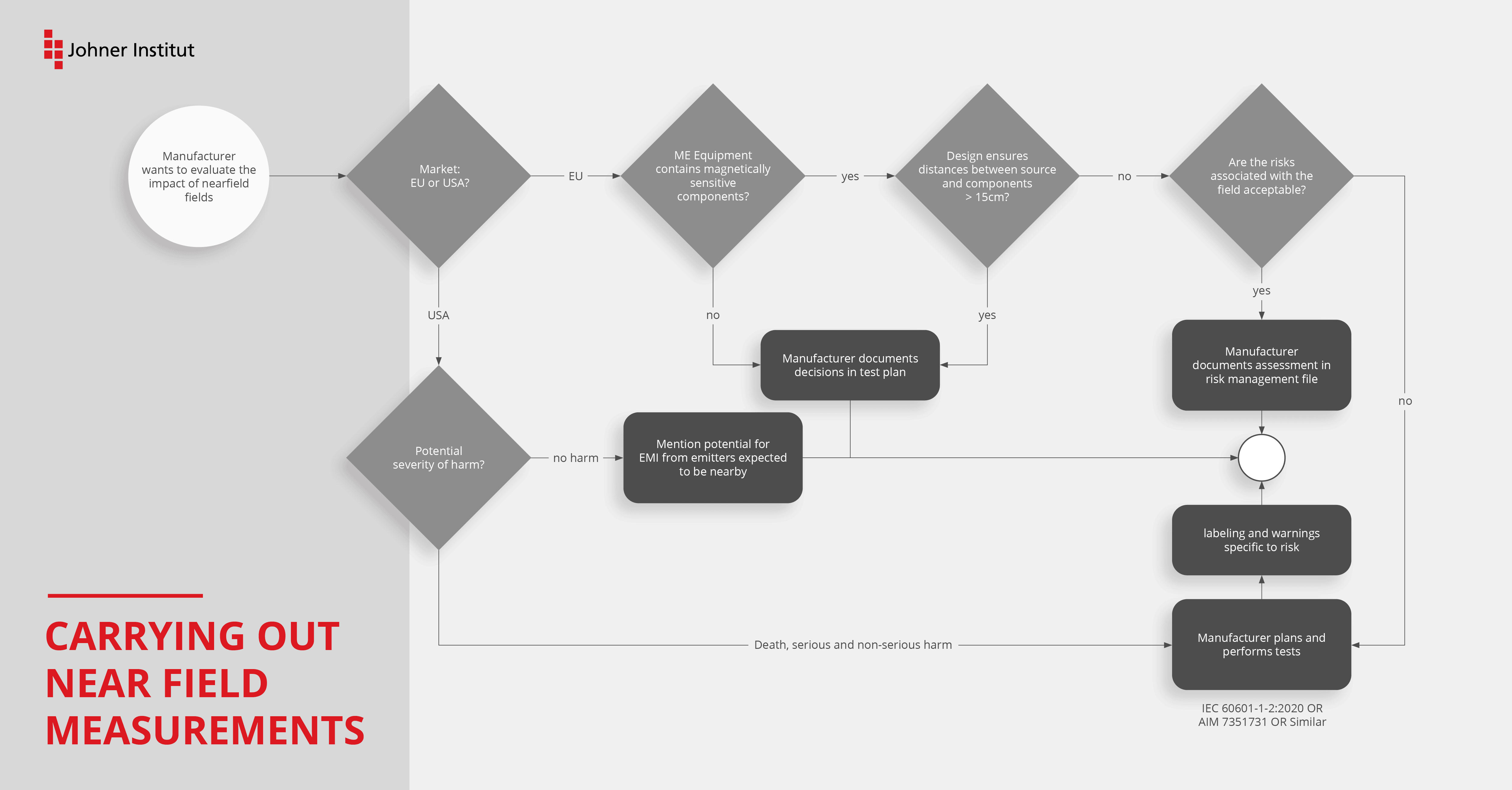
Hersteller müssen die Prüfung nach Tabelle 11 der Edition 4.1 für ME Geräte, Systeme und deren Zubehör in Betracht ziehen.
Die Prüfung muss dann nicht durchgeführt werden, wenn die Geräte
- keine störempfindlichen Bauteile oder Stromkreise besitzen oder
- deren Störung (bei Anwendung von Tabelle 11) nicht zum Verlust der Basissicherheit oder der wesentlichen Leistungsmerkmale führt oder
- bei denen der minimale Abstand von 15 cm zu einer Störquelle konstruktiv gewährleistet ist.
Den Ausschluss der Prüfung müssen Sie allerdings im EMV-Testplan begründen und dokumentieren.
Selbst wenn die neue Prüfung im Kapitel 8.11 für Ihr Gerät nicht durchzuführen ist (siehe Voraussetzungen), empfehlen wir die Prüfung dennoch. Damit lernen Sie Ihr Gerät besser kennen (insbesondere, wenn die Dokumentation dünn ist oder die ursprünglichen Entwickler nicht mehr da sind). Außerdem können Sie die „Design-Reserven“ ausloten, indem Sie die Prüfpegel z. B. noch deutlich über die der Tabelle 11 erhöhen. Mit den Ergebnissen erhalten Sie auch weitere Entscheidungskriterien für Ihre Produkt-Roadmap.
Tipp 6: Maßnahmen ergreifen und umsetzen
Erstellen Sie einen EMV-Testplan konform mit den Vorgaben der EN 60601-1-2 (4. Edition)
Führen Sie eine Gap-Analyse durch, um zu prüfen, ob Sie die zwischen den Versionen geänderten Anforderungen erfüllen. Das betrifft insbesondere die Anforderungen an das Gerätedesign und an die technische Dokumentation.
Prüfen Sie abhängig von den Ergebnissen Ihr Produkt und Ihre Dokumentation gemäß ER-Checkliste der MDD und GSPR-Checkliste der MDR auf Vollständigkeit und Korrektheit. Verwenden Sie dazu die Tabelle ZZ.1 im Anhang ZZ der EN-Version der Norm.
Leiten Sie aus den Ergebnissen der Punkte 2, 3, 7 und 8 eine Liste mit Maßnahmen für die Überarbeitung der Architektur (unter Einbezug der Risikoanalyse) ab.
Setzen Sie die Maßnahmen um und prüfen Sie Ihr Produkt in einem akkreditierten Prüflabor.
Erklären Sie die Konformität.
Nutzen Sie die Unterstützung des Johner Instituts
Das Johner Institut unterstützt Hersteller dabei, die Konformität ihrer Medizinprodukte und ihrer Dokumentation zu prüfen und diese bei Abweichungen mit schlanken und präzisen Maßnahmen wiederherzustellen. Dazu erstellt es Gap-Analysen, prüft Dokumente, erstellt Prüfpläne und hilft bei der Argumentation mit Behörden und Benannten Stellen.
Melden Sie sich bei uns (z. B. über das Kontaktformular), damit wir gemeinsam das konkrete Vorgehen festlegen und so die Konformität und Sicherheit Ihrer Produkte sicherstellen können.
6. Fazit, Zusammenfassung
Das Amendment 1 verbessert die bisherige vierte Edition der IEC 60601-1-2 und bildet gemeinsam mit ihr die Edition 4.1.
Diese Version der EMV-Norm ist eine konsequente Weiterentwicklung, die notwendig war, um technischen Trends Rechnung zu tragen und um die Verständlichkeit und Aktualität der Norm zu gewährleisten.
Für Hersteller verdeutlicht diese neue Ausgabe, dass sich der Stand der Technik ständig weiterentwickelt. Dem müssen sie begegnen. Sie sollten dabei aber immer risikobasiert vorgehen, um sowohl unnötige Aufwände als auch regulatorischen Ärger zu vermeiden.
Letztlich ist das Ziel, die Sicherheit und Leistungsfähigkeit von Medizinprodukten und damit die Sicherheit und Gesundheit von Patienten zu gewährleisten.
Änderungshistorie
- 2023-03-03: Entscheidungsdiagramm eingefügt
- 2021-03: Artikel aus dem Jahr 2016 völlig neu gestaltet


Hallo Herr Klessascheck,
stimmt Ihre Aussage im Kapitel „Harmonisierung und Übergangsfristen“, dass die benannten Stellen die Prüfberichte nach der 3. Edition der DIN EN 60601-1-2 akzeptieren und nur die Risikomanagementakte gepflegt/erweitert werden muss, wenn bereits auf dem Markt befindliche Geräte weiterhin unverändert produziert und vertrieben werden?
Dies hätte einen nicht zu unterschätzenden (positiven) Einfluss auf die zeitlichen und finanziellen Aufwände, die wir mit unserem recht großen Produktspektrum zu bewältigen haben, um die neue Ausgabe der 60601-1-2 auf diese „Altgeräte“ anzuwenden.
Freundliche Grüße
Ralf Philipp
Sehr geehrter Herr Prof. Johner,
wir sind Hersteller von Therapieliegen und möchten den Betrieb dieser Liegen, die über Netzleitungen mit einer Steckdose in einem Gebäude verbinden sind, über eine sogenannte „Sperrbox“ codiert unterbrechen.
D.h. durch Abziehen des Codestiftes aus der Sperrbox wird die Netzversorgung zur Therapieliege unterbrochen.
Durch diese Einrichtung wird sichergestellt, daß nur der Therapeut, der in Besitz des Codestiftes ist, kann die Stromversorgung wieder herstellen und die Therapieliege betreiben.
Jetzt fragen wir uns ob wir diese Sperrbox (LxBxH) 105x60x35mm, die fliegend in die Netzleitung zwischen Steckdose und elektrischem Höhenverstellantrieb fest verdrahtet eingebaut ist, überhaupt einer EMV-Prüfung unterzogen werden muß und wenn ja, wie diese geprüft werden muß.
Da wir als Hersteller und Inverkehrbringer der Therapieliegen nach dem Medizinproduktegesetz EN 60601-1 herstellen und CE-Zertifizieren, möchten wir das auch wir beim Einsatz der „Sperrbox“
an unseren Therapieliegen sicher stellen.
Können Sie uns da einen Tip geben?
Im Voraus vielen Dank für Ihre Hilfe.
Freundliche Grüße
Hubert Wassmer
Fa. HWK gGmbH