
Wer stoffliche Medizinprodukte wie Meerwasser-Nasensprays, schleimhautberuhigende Hustensäfte oder osmotisch wirksame Abführmittel herstellt, steht mit Inkrafttreten der Verordnung (EU) 2017/745 (MDR) vor gleich mehreren Herausforderungen:
- Die neue Regel 21 führt zu einer Höherklassifizierung von stofflichen Medizinprodukten der Klasse I. Das macht erstmalig die Einbindung einer Benannten Stelle erforderlich.
- Die MDR stellt höhere Anforderungen an die klinischen Daten. Diese betreffen auch den Nachweis der Äquivalenz.
Dieser Artikel zeigt Ihnen, was Sie tun können, um die regulatorischen Hürden zu meistern und die Marktfähigkeit Ihrer stofflichen Medizinprodukte auch über die Übergangsfristen hinaus zu gewährleisten. Das ist wichtig, um den Patienten weiterhin eine sichere und bewährte Gesundheitsversorgung zur Verfügung zu stellen.
Zudem liefert dieser Beitrag Ihnen Anhaltspunkte dafür, was Sie in Hinblick auf Neuentwicklungen oder signifikante Produktmodifikationen stofflicher Medizinprodukte beachten müssen (z. B. bei der Änderung von Rezeptur oder Zweckbestimmung), für die bereits ab dem Stichtag 26. Mai 2021 MDR-Compliance gefordert ist.
1. Einführung
a) Definition „Stoffliche Medizinprodukte“
b) Beispiele für stoffliche Medizinprodukte

Foto: Markus Heimbach
Zu den stofflichen Medizinprodukten gehören z. B.:
- Salzhaltige Nasentropfen oder -sprays
- Schleimhautbefeuchtende Sirupe, Rachensprays oder Lutschtabletten,
- Künstliche Tränenflüssigkeiten
- Vaginalcremes oder -gele zur Wiederherstellung des physiologischen Milieus
- Hautcremes oder -salben zur Befeuchtung/Abdichtung von Wunden, Narben oder bei Hauterkrankungen
- Orale Mittel zur Neutralisierung von Magensäure
- Osmotisch wirksame Abführmittel
- Produkte mit entschäumender Wirkung bei gasbedingten Magen-Darm Beschwerden
2. Herausforderungen für Hersteller stofflicher Medizinprodukte
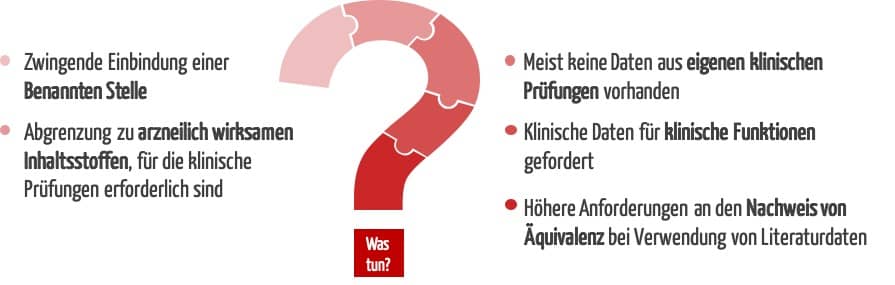
a) Pflicht zum Nachweis anhand klinischer Daten
Im Gegensatz zu vielen anderen Medizinprodukten haben stoffliche Medizinprodukte in der Regel klinische Funktionen wie die Reduktion von Symptomen wie z. B. Schnupfen, Husten, Bauchschmerzen oder Verstopfung und erzielen ihre Wirkung in unmittelbarem Kontakt mit dem menschlichen Körper. Daher können Sicherheit und Leistung bei diesen Produkten nicht oder nicht vollumfänglich anhand von präklinischen Daten oder Leistungsdaten belegt werden. Dieser Nachweis erfordert vielmehr klinische Daten.
Während klinische Prüfungen bei Klasse-III-Produkten und Neuentwicklungen in der Regel obligatorisch sind, wurden für lange auf dem Markt befindliche Bestandsprodukte mit geringem Risikoprofil unter der Richtlinie 93/42/EWG (MDD) meist keine eigenen klinischen Prüfungen durchgeführt.
Klinische Prüfungen sind mit erheblichem Zeit- und Ressourcenaufwand verbunden und stellen vor allem für kleinere mittelständische Unternehmen große Herausforderungen dar.
b) Höherklassifizierung stofflicher Medizinprodukte & Einbindung Benannter Stellen
Die MDR hat für stoffliche Medizinprodukte in Anhang VIII als neue Klassifizierungsregel die Regel 21 eingeführt:
„Produkte, die aus Stoffen oder Kombinationen von Stoffen bestehen, die dazu bestimmt sind, durch eine Körperöffnung in den menschlichen Körper eingeführt oder auf die Haut aufgetragen zu werden, und die vom Körper aufgenommen oder lokal im Körper verteilt werden, werden wie folgt zugeordnet:
MDR Anhang VIII, Regel 21
– der Klasse III, wenn sie oder ihre Metaboliten systemisch vom menschlichen Körper aufgenommen werden, um ihre Zweckbestimmung zu erfüllen;
– der Klasse III, wenn sie ihre Zweckbestimmung im Magen oder im unteren Magen-Darm-Trakt erfüllen und wenn sie oder ihre Metaboliten systemisch vom menschlichen Körper aufgenommen werden;
– der Klasse IIa, wenn sie auf die Haut aufgetragen werden oder in der Nasenhöhle oder der Mundhöhle bis zum Rachen angewandt werden und ihre Zweckbestimmung an diesen Höhlen erfüllen und
– der Klasse IIb in allen anderen Fällen“
Damit sind alle stofflichen Medizinprodukte, die unter der MDD bislang in Klasse I fielen, mit der MDR von einer Höherklassifizierung betroffen. Für die weitere Marktfähigkeit über die Übergangsfristen hinaus wird erstmalig die Einbindung einer Benannten Stelle erforderlich.
Lesen Sie hier mehr zur Klassifizierung von Medizinprodukten und zu den Übergangsfristen.
c) Pflicht zu klinischen Prüfungen bei bestimmten stofflichen Medizinprodukten
Der Schlüssel für die Unterscheidung zwischen Arzneimitteln und stofflichen Medizinprodukten liegt in der korrekten Interpretation des Wirkmechanismus, mit dem die Produkte ihre bestimmungsgemäße Hauptwirkung erzielen. Während Arzneimittel eine pharmakologische, immunologische oder metabolische Wirkweise haben, sind stoffliche Medizinprodukte über ihren chemischen und/oder physikalischen Mechanismus definiert, über den sie ihre Hauptwirkung erzielen. Dies spiegelt sich auch in ihrer Zweckbestimmung wider.
Wenn stoffliche Medizinprodukte arzneilich wirksame Stoffe enthalten, die die bestimmungsgemäße Hauptwirkung unterstützen, werden sie nach Regel 14 MDR der Klasse III zugeordnet. Für diese Produkte sind, wie für alle anderen stofflichen Medizinprodukte der Klasse III, eigene klinische Prüfungen obligatorisch.
d) Höhere Anforderungen an den Nachweis der Äquivalenz
Laut MDR kann eine rein auf Literatur gestützte klinische Bewertung nur durchgeführt werden, wenn eine ausreichende Äquivalenz mit einem Referenzprodukt vorliegt. Der Nachweis der Äquivalenz ist mit der Richtlinie MEDDEV 2/7.1 rev. 4 und nun auch mit der MDR deutlich verschärft worden gegenüber den Vorgaben der MDD.
Laut ANNEX XIV Teil A der MDR werden zum Nachweis der Äquivalenz die folgenden technischen, biologischen und klinischen Merkmale herangezogen:
Klinische Äquivalenz
Das Produkt wird
- unter der gleichen klinischen Bedingung oder zum gleichen klinischen Zweck (einschließlich eines ähnlichen Schweregrads und Stadiums der Krankheit),
- an der gleichen Körperstelle und
- bei ähnlichen Patientenpopulationen angewandt,
- hat die gleichen Anwender und
- erbringt eine ähnliche, maßgebliche und entscheidende Leistung im Hinblick auf die erwartete klinische Wirkung für eine spezielle Zweckbestimmung.
Technische Äquivalenz
Das Produkt
- ist von ähnlicher Bauart,
- wird unter ähnlichen Anwendungsbedingungen angewandt,
- hat ähnliche Spezifikationen und Eigenschaften,
- verwendet ähnliche Entwicklungsmethoden und
- hat ähnliche Funktionsgrundsätze und entscheidende Leistungsanforderungen.
Biologische Äquivalenz
Das Produkt
- verwendet die gleichen Materialien oder Stoffe im Kontakt mit den gleichen menschlichen Geweben oder Körperflüssigkeiten
- für eine ähnliche Art und Dauer des Kontakts
- bei ähnlichem Abgabeverhalten der Stoffe.
In der MDR heißt es dazu weiter, dass die Inhaltsstoffe in einer Weise gleichartig sein müssen, dass es keinen klinisch bedeutsamen Unterschied bei der Sicherheit und klinischen Leistung gibt. Weiterhin ist es erforderlich, dass sich die Bewertung der Äquivalenz auf eine angemessene wissenschaftliche Begründung stützt.
Beachten Sie, dass das Medizinprodukt und das Äquivalenzprodukt bei einigen Attributen identisch sein müssen, während sie bei anderen nur „ähnlich“ sein dürfen.
Lesen Sie hier mehr zur Äquivalenz von Medizinprodukten.
e) Schwierigkeiten beim Zugang zu Daten der Äquivalenzprodukte
Die MDR fordert von den Herstellern, eindeutig nachzuweisen, dass sie über einen hinreichenden Zugang zu den Daten der Äquivalenzprodukte verfügen, die nötig sind, um die Äquivalenz zu belegen.
„Es muss eindeutig nachgewiesen werden, dass die Hersteller über einen hinreichenden Zugang zu den Daten von Produkten, mit denen sie die Gleichartigkeit geltend machen, verfügen, um die von ihnen behauptete Gleichartigkeit belegen zu können.“
MDR Anhang XIV
Für implantierbare Produkte und Klasse-III-Produkte gilt dabei:
„Die beiden Hersteller haben einen Vertrag geschlossen, in dem dem Hersteller des zweiten Produkts ausdrücklich der uneingeschränkte Zugang zur technischen Dokumentation durchgängig gestattet wird“.
MDR Artikel 61 (5)
Die Leitlinie MDCG 2020-5 führt an, dass für alle anderen Produkte ein entsprechender Vertrag nicht erforderlich ist:
“For devices other than implantable devices and class III devices there is no MDR requirement of a contract between the manufacturers for regulating the access to the technical documentation.”
MDCG 2020-5
Die Anforderungen der MDR nach einem „hinreichenden Zugang“ zu den Daten der Äquivalenzprodukte und dem „ausreichenden klinischen Nachweis“ sind die am stärksten auslegungsbedürftigen. Obwohl und weil noch nicht ganz klar ist, wie die Benannten Stellen diese Begriffe im Einzelnen auslegen werden, sind Sie als Hersteller bereits jetzt gefordert, darzulegen und zu begründen, was Sie in Bezug auf Ihre stofflichen Medizinprodukte als ausreichend bzw. hinreichend erachten.
f) Ungenaue Festlegung zum Umfang klinischer Daten
Während der Nachweis von klinischer Leistung und Sicherheit und der klinische Nutzen von stofflichen Medizinprodukten anhand klinischer Daten erfolgen muss, richtet sich der Umfang, der als ausreichend angesehen werden kann, nach den Eigenschaften des Produkts und seiner Zweckbestimmung:
„Der Hersteller spezifiziert und begründet den Umfang des klinischen Nachweises, der erforderlich ist, um die Erfüllung der einschlägigen grundlegenden Sicherheits- und Leistungsanforderungen zu belegen. Der Umfang des klinischen Nachweises muss den Merkmalen des Produkts und seiner Zweckbestimmung angemessen sein.“
MDR Artikel 61(1)
Der Artikel zu den klinischen Daten erläutert, was klinische Daten sind und wie diese erhoben werden können.
g) Neue Anforderungen an die Dokumentation der Kinetik der Inhaltsstoffe
Eine weitere, mit der MDR neu gestellte Anforderung an stoffliche Medizinprodukte, die vom Körper aufgenommen oder lokal im Körper verteilt werden und eine Arzneimittelkomponente enthalten, ist die Dokumentation der Kinetik dieser Inhaltsstoffe Dabei geht es um die Dokumentation der Absorption, Verteilung, Verstoffwechselung und Ausscheidung, ähnlich wie sie für Humanarzneimittel gefordert ist.
„Produkte, die aus Stoffen oder Kombinationen von Stoffen bestehen, die dazu bestimmt sind, in den menschlichen Körper eingeführt zu werden, und die vom Körper aufgenommen oder lokal im Körper verteilt werden, müssen gegebenenfalls und beschränkt auf die nicht unter diese Verordnung fallenden Aspekte die in Anhang I der Richtlinie 2001/83/EG festgelegten Anforderungen erfüllen in Bezug auf die Bewertung von Resorption, Verteilung, Metabolismus, Ausscheidung, lokale Verträglichkeit, Toxizität, Wechselwirkungen mit anderen Medizinprodukten, Arzneimitteln oder sonstigen Stoffen sowie mögliche unerwünschte Reaktionen gemäß dem nach dieser Verordnung geltenden Konformitätsbewertungsverfahren.“
MDR Anhang I 12.2
Stellen Sie daher sicher, dass die Kinetik der Inhaltsstoffe in Ihrer Biokompatibilitätsbewertung dokumentiert ist.
3. Praxistipps
Um Ihre stofflichen Bestandsprodukte auch nach dem Ende der Übergangsfristen weiter vermarkten zu dürfen, empfehlen wir Ihnen die Umsetzung folgender Schritte:
a) Prüfen Sie den Wirkmechanismus der Inhaltsstoffe Ihres stofflichen Medizinprodukts
Gleichen Sie den Wirkmechanismus mit der Rezeptur und der Zweckbestimmung ab. Nur bei einem physikochemischen und/oder physikalischen Mechanismus aller für die Zweckbestimmung verantwortlichen Inhaltsstoffe können Sie eine eindeutige Abgrenzung zu den arzneilichen Wirkungen vornehmen und sicherstellen, dass Ihr stoffliches Medizinprodukt nicht unbeabsichtigt in Klasse III eingestuft wird. Für Produkte dieser Klasse fordert die MDR klinische Prüfungen.
Während einige Substanzen einen eindeutig bestimmbaren Wirkmechanismus haben, kann z. B. bei pflanzlichen Stoffgemischen die Art der Aufbereitung eine entscheidende Rolle für den resultierenden Mechanismus spielen. Für andere Inhaltsstoffe existiert ein Schwellenwert für die pharmakologische Wirkung, den Sie in Ihrer Rezeptur berücksichtigen sollten. Sprechen Sie uns einfach an, wenn Sie Hilfe bei der Bestimmung des Wirkmechanismus oder der Abgrenzung zu den Arzneimitteln benötigen.
b) Sichten Sie vorhandene (klinische) Daten
Sichten Sie vorhandene (klinische) Daten aus den folgenden Quellen:
- Prüfungen mit dem eigenen Produkt / Studien mit Äquivalenzprodukten aus der Literatur
Prüfen Sie die Äquivalenz bei Verwendung von Literaturdaten entsprechend den Anforderungen von MDR und dem MDCG 2020-5 Dokument zur klinischen Bewertung und Äquivalenz. Dokumentieren Sie, dass Sie über einen hinreichenden Zugang zu den Daten der Äquivalenzprodukte verfügen und Ihnen genügend Daten vorliegen, um eine Äquivalenzbewertung vornehmen zu können. - Daten aus der Post-Market Surveillance (PMS)
Diese klinischen Daten können bei ausreichendem Vermarktungszeitraum und etabliertem Qualitätsmanagementsystem dazu beitragen, die Sicherheit Ihres stofflichen Bestandsprodukts zu belegen. - Allgemeine PMCF-Aktivitäten
Zu den im PMCF-Plan und -Bericht aufgeführten allgemeinen PMCF-Aktivitäten, die mit der MDR für jedes Produkt gefordert sind, gehören z. B. das Zusammenführen klinischer Erfahrungen, die Einholung eines Anwenderfeedbacks sowie die Durchsicht wissenschaftlicher Fachliteratur und anderer Quellen. Die Auswertung dieser Aktivitäten liefert weitere klinische Daten für Ihr Produkt.
c) Identifizieren Sie fehlende Daten anhand einer Gap-Analyse
Legen Sie fest, welche Funktionen Ihres stofflichen Medizinprodukts anhand klinischer Daten und welche anhand von Leistungsdaten nachgewiesen werden können. Spezifizieren Sie vor dem Hintergrund der Eigenschaften des Produkts und seiner Zweckbestimmung den Umfang an klinischen Daten, den Sie für Ihr stoffliches Bestandsprodukt als ausreichend erachten.
d) Erzeugen Sie fehlende präklinische/In-vitro-Daten zum Nachweis der Biokompatibilität und/oder nicht-klinischer Leistungsfunktionen/Claims
Wenn präklinische Daten oder In-vitro-Daten geeignet sind, bestehende Lücken zum Nachweis der Sicherheit und Leistung ihres stofflichen Medizinprodukts zu schließen, sollten Sie diese jetzt generieren.
e) Erzeugen Sie fehlende klinische Daten für Ihr stoffliches Bestandsprodukt über eine PMCF-Studie, falls erforderlich
Es gibt es nach der Leitlinie MDCG 2020-6 zur klinischen Evidenz einen Ermessensspielraum dafür, ob bzw. in welchen Fällen die Durchführung von PMCF-Studien für Bestandsprodukte erforderlich sind:
“In some cases, it may be necessary for the manufacturer to undertake PMCF to generate new data for these legacy devices prior to CE marking under the MDR, whereas in other cases (…) it may be possible to demonstrate conformity with the relevant GSPRs with a more limited clinical data set.”
MDCG 2020-6
Wenn klinische Daten erforderlich sind, um bestehende Lücken zu schließen, sollten Sie eine PMCF-Studie immer dann initiieren, wenn
- die vorhandenen klinischen Daten zum Nachweis von Sicherheit, Leistung und klinischem Nutzen inkl. Claims für die klinischen Funktionen nicht ausreichen,
- der Nachweis der Leistung und/oder der Sicherheit ausschließlich auf präklinischen Daten beruht oder
- es aus klinischen/präklinischen Daten des eigenen Produkts oder von Äquivalenzprodukten, aus dem Risikomanagement oder aus dem State of the Art Anhaltspunkte dafür gibt, dass Sicherheit, Leistung und/oder der klinische Nutzen hinsichtlich bestimmter Aspekte eine Neubewertung erfordern.
Maike Andersson erläutert im Gespräch mit Professor Johner, was stoffliche Medizinprodukte sind und wie sich diese von Arzneimitteln abgrenzen. Sie listet die (neuen) Anforderungen an diese Klasse an Medizinprodukten und gibt Tipps, wie Hersteller diese erfüllen kommen.
Diese und weitere Podcast-Episoden finden Sie auch hier.

4. Fazit und Zusammenfassung
Um Ihre stofflichen Bestandsprodukte unter der MDR auch nach dem Ende der Übergangsfristen weiter vermarkten zu dürfen, raten wir Ihnen, jetzt zu handeln, um die regulatorischen Anforderungen erfüllen zu können.
Nutzen Sie die Zeit und erzeugen Sie fehlende (klinische) Daten. Der Aufwand sollte sich nach der Risikoklasse richten und gleichzeitig Ressourcen schonen, d. h. so gering wie möglich und so hoch wie nötig sein, um die Patientensicherheit zu gewährleisten. Die verschiedenen Möglichkeiten zur Generierung von Daten für stoffliche Bestandsprodukte in Abhängigkeit von der Risikoklasse sind in Abb. 2 zusammengefasst.
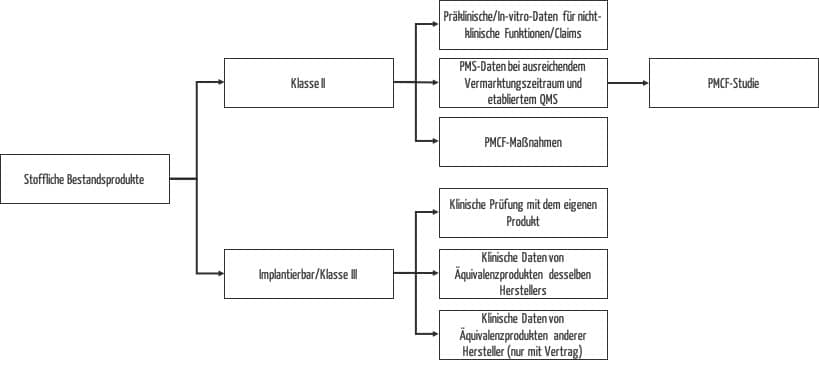
Während die Durchführung von klinischen Prüfungen für Klasse-III-Produkte und Neuentwicklungen obligatorisch ist, stehen Ihnen für alle anderen stofflichen Bestandsprodukte einfache, effektive und ressourcenschonende Möglichkeiten zur Erzeugung klinischer Daten zur Verfügung. Nutzen Sie die Chance und starten Sie für diese Produkte PMCF-Studien, wenn die vorhandenen klinischen Daten zum Nachweis von Sicherheit und Leistung der klinischen Funktionen nicht ausreichen.
Das Johner Institut unterstützt Sie darin, die regulatorischen Hürden zu überwinden und die Marktfähigkeit Ihrer stofflichen Medizinprodukte sicherzustellen. Wir beantworten Fragen wie:
- Ist Ihr stoffliches Produkt überhaupt ein Medizinprodukt? Falls ja, in welche Klasse fällt es?
- Enthält Ihr stoffliches Medizinprodukt Stoffe, die möglicherweise als Arzneimittel gelten?
- Welche klinischen Daten benötigen Sie für die klinische Bewertung Ihres stofflichen Medizinprodukts? Wie können Sie fehlende Daten am schnellsten erheben?
- Erfüllt Ihr Medizinprodukt die Anforderungen der MDR? Dürfen Sie Ihr bestehendes Produkt weiterhin gesetzeskonform vermarkten?
Nehmen Sie Kontakt auf oder nutzen Sie bei kurzen Fragen gerne das kostenlose Micro-Consulting des Johner Instituts. Gerne erstellen unsere Experten:innen auch eine klinische Bewertung oder Biokompatibilitätsbewertung für Ihre stofflichen Medizinprodukte.


Zur Definition von Äquivalenz bzw. Gleichartigkeit:
Welche stofflichen Medizinprodukte gelten als so „ähnlich“, um mit einem Produkt eine klinische Prüfung durchzuführen, und dann die anderen Produkte darauf für die klinische Bewertung zu beziehen? Wie „gleichartig“ müssen Produkte sein, um sich auf die klinischen Daten eines gleichartigen Produktes beziehen zu können? Wann ist „in einer Weise gleichartig sein, dass es keinen klinisch bedeutsamen Unterschied bei der Sicherheit und klinischen Leistung der Produkte“ dieser Punkt erfüllt?
– bzw. welche sind so „verschieden“, dass eigene klinische Daten/Studien notwendig werden?
Welche Rolle spielen verschiedene Hilfsstoffe bei der Definition der „Gleichartigkeit“?
• Gibt es hierzu Empfehlungen durch europäische Guidelines oder veröffentlichte Fallentscheidungen?
Beispiele:
1. a) Nasenspray mit 0.9% NaCl
b) Nasenspray mit 0.9% NaCl sowie Zitronenaroma
c) Nasenspray mit 0.9% NaCl sowie Salbeiaroma
2. a) Nasenspray mit 0.9% NaCl, konserviert
b) Nasenspray mit 0.9% NaCl, konservierungsmittelfrei
3) a) Nasenspray mit isotoner Meersalzlösung (7 Komponenten)
b) Nasenspray mit isotoner Meersalzlösung (7 Komponenten, anderes Mengenverhältnis als bei a) )
c) Nasenspray mit isotoner Meersalzlösung (4 Komponenten)
Liebe Frau Linnemayr,
vielen Dank für Ihre Frage.
Ich gehe einmal davon aus, dass die technische und klinische Äquivalent innerhalb der Produktgruppen 1, 2 und 3 gegeben ist.
Bei den Nasensprays mit den verschiedenen Aromakomponenten (1 a und b) bzw. mit oder ohne Konservierungsstoffen (2 a und b) würde ich von einer biologischen Gleichwertigkeit ausgehen.
Guidelines oder Empfehlungen dazu sind mir nicht bekannt, aber ich würde hier argumentieren, dass es sich um standardisierte Zusätze handelt, die nicht für die Zweckbestimmung verantwortlich sind und deren Sicherheitsprofil bekannt ist. Hier könnten Sie auch auf die Biokompatibilitätsbewertung verweisen. Ähnliches gilt aus meiner Sicht auch für Hilfsstoffe nach aktueller Pharm. Eur.
Bei den isotonischen Sprays mit qualitativen und quantitativen Unterschieden bezüglich der Zusammensetzung der für die Zweckbestimmung verantwortlichen Inhaltsstoffe lässt sich Ihre Frage leider nicht so schnell beantworten. Hier sehe ich „Gaps“, die sie schließen sollten. Möglicherweise bietet sich an, eine in-vitro Studie durchzuführen, um die biologische Äquivalenz nachzuweisen, wenn eine solche hier gegeben ist, z. B. weil ausschließlich der isotone Charakter der Lösung für die Sicherheit und klinische Leistung der Produkte entscheidend ist.
Ich hoffe, ich konnte Ihre Fragen hiermit hinreichend beantworten.
Viele Grüße,
Maike Andersson
Danke!
Guten Tag Frau Andersson,
meine Frage bezieht sich auf die oben erwähnte Klassifizierungsregel 21, dritter Spiegelstrich:
– der Klasse IIa, wenn sie auf die Haut aufgetragen werden oder in der Nasenhöhle oder der Mundhöhle bis zum Rachen angewandt werden und ihre Zweckbestimmung an diesen Höhlen erfüllen…
Verstehe ich das richtig, dass die Aussage „bis zum Rachen“, den gesamten Rachen miteinschließt und dieses ebenfalls als Höhle für die Erfüllung der Zweckbestimmung versteht?
Vielen Dank im Voraus und viele Grüße,
Ilona Milke
Liebe Frau Milke,
vielen Dank für Ihre interessante Frage, die schon häufiger an mich herangetragen wurde. Vieles in der MDR ist etwas missverständlich formuliert, so auch dieser Punkt in der Regel 21. Sie haben es genau richtig erkannt:
Wenn das Produkt seine Zweckbestimmung im Rachen erfüllt, ist der dritte Spiegelstrich anzuwenden, d.h. dass der Rachen selbst als Höhle hier miteingeschlossen ist.
Viele Grüße,
Maike Andersson
Guten Tag Frau Andersson,
eine kleine Frage zur Kinetik (Punkt 2g): Sie schreiben, dass die Daten zur Kinetik nur für Medizinprodukte erhoben werden müssen, die eine Arzneimittelkomponente haben („die vom Körper aufgenommen oder lokal im Körper verteilt werden und eine Arzneimittelkomponente enthalten,“), allerdings lese ich aus der MDR (Anhang I, 12 und Anhang IX 5.4a) Folgendes:
„Produkte, zu deren Bestandteilen ein Stoff gehört, der als Arzneimittel gilt, und Produkte, die aus Stoffen oder aus Kombinationen von Stoffen bestehen, die vom menschlichen Körper aufgenommen oder lokal im Körper verteilt werden“ bzw. „Die Qualität und Sicherheit von Produkten, die aus Stoffen oder Kombinationen von Stoffen bestehen, die dazu bestimmt sind, über eine Körperöffnung in den menschlichen Körper eingeführt oder auf die Haut aufgetragen zu werden, und die vom Körper aufgenommen oder lokal im Körper verteilt werden, werden“.
Meine Interpretation davon wäre, dass die Daten zur Kinetik für alle stofflichen Medizinprodukte gefordert sind, die in den Körper eingeführt / auf die Haut aufgetragen werden und sich lokal verteilen (z.B. auch ein Meerwasser-Nasenspray).
Verstehe ich hier grundsätzlich etwas falsch und es geht doch nur um Produkte mit AM-Komponente, oder sind tatsächlich klinische Daten erforderlich?
Und falls ja, gibt es Leitlinien zur Umsetzung der genannten Arzneimittelrichtlinie?
Vielen Dank im Voraus und beste Grüße
Sehr geehrter Herr Faller,
hiermit sind nur Produkte gemeint, die aus Stoffen oder Kombinationen von Stoffen bestehen, die dazu bestimmt sind, in den menschlichen Körper eingeführt zu werden, und die vom Körper aufgenommen oder lokal im Körper verteilt werden. Entscheidend hierfür ist die Formulierung der Zweckbestimmung und die Inhaltsstoffe des Produktes. Bei diesen Produkten müssen die entsprechenden Nachweise zur Kinetik mittels vorklinischer und/oder klinischer Daten erfolgen (siehe Anhang II, 6.2).
Arzneimittel wirken pharmakologisch, immunologisch oder metabolisch. Wenn stoffliche Medizinprodukte arzneilich wirksame Stoffe enthalten, die die bestimmungsgemäße Hauptwirkung unterstützen, werden sie nach Regel 14 MDR der Klasse III zugeordnet. Für diese Produkte sind, wie für alle anderen stofflichen Medizinprodukte der Klasse III, eigene klinische Prüfungen obligatorisch.
Unser Fokus liegt im Medizinproduktebereich und die Interpretation der entsprechenden Leitlinien.
Viele Grüße,
Nadine Jurrmann.
Sehr geehrte Frau Jurrmann,
hier muss ich doch nochmal nachhaken. Sie sprechen von stofflichen Medizinprodukten mit Arzneimittelkomponente und stofflichen Medizinprodukten der Klasse III.
In der MDR steht „Produkte, zu deren Bestandteilen ein Stoff gehört, der als Arzneimittel gilt, UND Produkte, die aus Stoffen oder aus Kombinationen von Stoffen bestehen, die vom menschlichen Körper aufgenommen oder lokal im Körper verteilt werden“. Das beinhaltet meiner Meinung nach auch einige stoffliche Medizinprodukte der Klasse IIb. Beispiel: Meerwasser-Panthenol-Nasenspray -> besteht aus Kombination von Stoffen, wird teilweise resorbiert und verteilt sich auf jeden Fall lokal im menschlichen Körper.
Für die Erhebung der pharmakokinetischen Daten soll entsprechend der EU-Richtlinie 2001/83/EG gearbeitet werden – das ist eine Arzneimittelrichtlinie, die im Falle von stofflichen Medizinprodukten jetzt auch für MP-Hersteller Anwendung findet. Gibt es hierzu Arbeitsdokumente speziell für Medizinprodukte-Hersteller (da jetzt wohl einige Hersteller betroffen sein müssten).
Daher die ursprüngliche Frage: ist es überhaupt noch möglich, solche Daten für stoffliche Medizinprodukte nicht zu erheben (hierbei geht es mir speziell um stoffliche Medizinprodukte, die weder der Klasse III angehören noch eine Arzneimittelkomponente haben)?
Danke und viele Grüße
Sehr geehrter Herr Faller,
vielen Dank für Ihre spannenden Fragen. Der Umfang übersteigt leider dem, was wir kostenlos anbieten können. Ich antworte Ihnen dennoch gerne kurz zu jeder Frage:
1. Im Moment sind uns keine Arbeitsdokumente speziell für Medizinprodukte-Hersteller bekannt. Jedoch sind Leitlinien der MDCG zum Thema Borderline & Klassifizierung geplant.
2. Wenn es sich um standardisierte Zusätze handelt, die nicht für die Zweckbestimmung verantwortlich sind, sollten die vorklinischen und klinischen Daten, die mit jedem Medizinprodukt erhoben werden, ausreichen.
Weitere Fragen könnten im Rahmen unserer kostenpflichtigen Dienstleistungen beantwortet werden.
Viele Grüße,
Nadine Jurrmann.
Sehr geehrte Frau Andersson,
Vielen Dank für den sehr interessant Beitrag. Ist er auch in englischer Sprache verfügbar?
Mit besten Grüssen,
Dr. caludia Mattern
Liebe Frau Dr. Mattern,
den englischen Beitrag finden Sie unter folgendem Link: https://www.johner-institute.com/articles/product-development/and-more/substance-based-medical-devices
Herzliche Grüße
Anja Segschneider | Redaktion
Guten Tag Herr Faller,
ich freue mich sehr, von Ihnen zu hören und hoffe, dass Sie meine Antwort auf diesem Wege erreicht.
Die Forderung nach der Dokumentation der Kinetik der Inhaltsstoffe von stofflichen Medizinprodukten erzeugt eine große Unsicherheit.
Unter 2g) zitiere ich den Anhang I Abschnitt 12.2 der MDR. Ein entscheidender Halbsatz in dem betreffenden Absatz lautet:“ beschränkt auf die nicht unter diese Verordnung fallenden Aspekte…“.
Dies interpretiere ich so, dass die Dokumentation lediglich für den arzneilichen Anteil eines Klasse III Medizinproduktes gefordert ist, der nicht unter die MDR fällt. Eine Dokumentation der Pharmakokinetik von stofflichen Medizinprodukten ohne Komponente mit pharmakologischen, immunologischen oder metabolischem Wirkmechanismus würde aus meiner Sicht keinen Sinn ergeben, da sie zwar vom Körper aufgenommen oder lokal im Körper verteilt werden können, aber nicht verstoffwechselt werden.
Viele Grüße,
Maike Andersson
Sehr geehrte Frau Andersson,
könnten Sie mir bei der Fragestellung helfen, wie es sich bei stofflichen Medizinprodukten in Bezug auf die Anforderungen an die Reinraumklasse verhält?
Hätten Sie da vielleicht einen Hinweis für mich, was dabei zu beachten wäre und welche Regelwerke und Normen hierfür relevant sein könnten?
Vielen herzlichen Dank im Voraus.
Mit freundlichen Grüßen
Ilona Milke
Sehr geehrte Frau Milke,
vielen Dank für Ihre interessante Frage!
Alle regulatorischen Anforderungen bzgl. der Tätigkeiten im Reinraum werden in der Norm DIN ISO 14644 abgebildet. Sie besteht aus 18 Kapiteln und deckt Fragen bzgl. Betrieb, Klassifizierung, Oberflächenreinheit, Partikelgrößen etc. ab.
Für ihre Frage der Stofflichen Medizinprodukte helfen die folgenden beiden Kapitel:
Kapitel 14644-14 „Bewertung der Reinraumtauglichkeit von Geräten durch Partikelkonzentrationen in der Luft“
Kapitel 14644-15 “ Bewertung der Reinraumtauglichkeit von Ausrüstungsgegenständen und Materialien anhand der chemischen und Oberflächenkonzentration“
Wenn es um den Betrieb des Reinraums geht ist die DIN 12599 maßgeblich, Sie können aber auch Informationen aus der Leitlinie VDI 2083 gewinnen, die das Arbeiten und den Betrieb von Reinräumen reguliert.
Ich hoffe, Ihre Frage damit hinreichend beantwortet zu haben.
Mit freundlichen Grüßen
Daniel Schulke
Guten Tag Frau Andersson,
Vielen Dank für den interessanten Beitrag!
Können Sie mir bitte Ihre Interpretation des Nebensatzes in der Klassifizierungsregel 21 geben?
„…, und die vom Körper aufgenommen oder lokal im Körper verteilt werden,…“
Für mich sieht das aus nach einer notwendigen Bedingung („UND“) für die Anwendbarkeit der Regel.
Das würde bedeuten, dass es auch stoffliche Medizinprodukte gibt (nämlich die die nicht vom Körper aufgenommen oder lokal im Körper verteilt werden), wofür Regel 21 keine Relevanz hat.
Allerdings differenzieren Sie hier in Ihrem Beitrag nicht und weiten u.a. die Implikationen dieser Klassifizierungsregel für früheren Klasse I stoffliche Medizinprodukte auf „ALLE“ stoffliche Medizinprodukte aus:
„Damit sind alle stofflichen Medizinprodukte, die unter der MDD bislang in Klasse I fielen, mit der MDR von einer Höherklassifizierung betroffen…“. Die Differenzierung nach Aufnahme vom/Verteilung im Körper oder nicht, wurde dabei also ignoriert.
Wenn Regel 21 jedoch tatsächlich ALLE stoffliche MP betrifft, warum wurde dieser Zusatz in der Regel 21 überhaupt aufgenommen oder wie ist es anders zu verstehen?
Vielen Dank im Voraus und beste Grüße
Sehr geehrter Herr Vander Beken,
Laut MDCG 2021-24 möchte ich Ihnen folgende Erläuterung geben:
Die Regel 21 deckt ein breites Spektrum von Medizinprodukten ab, die ausschließlich aus Stoffen bestehen. In diesem Zusammenhang bedeutet „Stoff“ jeden Stoff, der Teil des Medizinprodukts ist, einschließlich derjenigen, die der Definition von „Stoff“ in Artikel 1 Absatz 3 der Richtlinie 2001/83/EG entsprechen. Dies gilt unter der Voraussetzung, dass sie nicht durch die MDR ausgeschlossen sind (z.B. Ausschlusskriterien in Artikel 1 (6) (h)). Der spezifische medizinische Zweck wird vom Hersteller unter den in Artikel 2 Absatz 1 der MDR aufgeführten Punkten angegeben.
Wichtig ist, dass, wenn die bestimmungsgemäße Hauptwirkung solcher stoffgebundenen Medizinprodukte durch pharmakologische, immunologische oder metabolische Mittel erreicht oder unterstützt wird, die Richtlinie 2001/83/EG oder die Verordnung (EG) Nr. 726/2004 bzw. Regel 14 auf das Produkt anzuwenden ist.
Bei der Anwendung der Regel 21 und der Einstufung (Klassifizierung) werden der Ort der Anwendung des Medizinprodukts sowie der Ort, an dem das Medizinprodukt seine Wirkung im oder am menschlichen Körper entfaltet, berücksichtigt. Für die Zwecke dieser Regel werden auch Nägel als „Haut“ betrachtet.
Die Hersteller von Produkten auf Stoffbasis sollten eindeutige Informationen zur Verfügung stellen, die die Wirkungsweise belegen, durch die der Stoff den beabsichtigten spezifischen medizinischen Zweck erreicht, als Grundlage für die Anwendung dieser Regel, einschließlich des Ortes der Anwendung sowie des Ortes, an dem die Wirkung im oder am Körper erzielt wird.
Mit freundlichen Grüßen,
Nadine Jurrmann.