
Die Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAMIV) löst die Medizinprodukte-Sicherheitsplanverordnung (MPSV) ab.
Welche neuen Anforderungen der MPAMIV betreffen die Hersteller und Betreiber? Welche Auswirkungen haben diese auf deren internen Vorgaben, z. B. deren QM-Systeme?
Die Kenntnis der regulatorischen Vorgaben ist wichtig, denn beim Meldewesen zeigen sich Behörden wenig kulant.
Aber es gilt nicht nur, empfindliche Geldbußen zu vermeiden, sondern auch, die Sicherheit der Patienten zu gewährleisten.
1. Weshalb es der MPAMIV bedarf
Das bisherige Medizinproduktegesetz MPG verpflichtet die Hersteller (bzw. deren Sicherheitsbeauftragte) „bekannt gewordene Meldungen über Risiken bei Medizinprodukten zu sammeln, zu bewerten und die notwendigen Maßnahmen zu koordinieren“. Details regelt die Medizinprodukte-Sicherheitsplanverordnung.
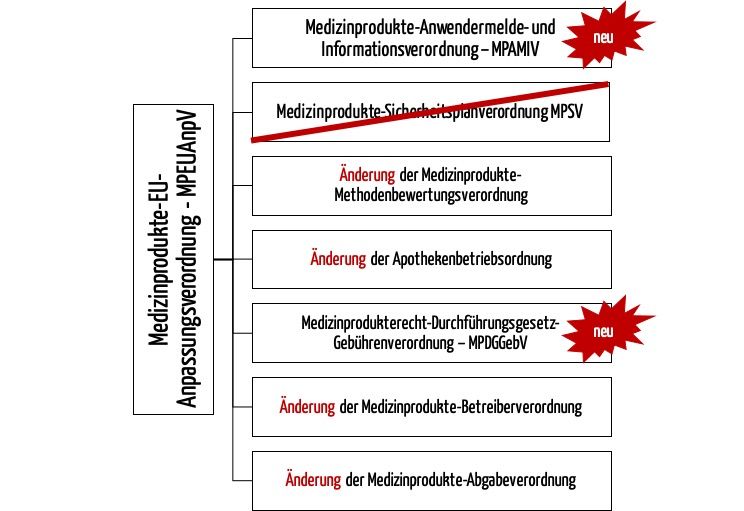
Weil die EU-Verordnungen (MDR, IVDR) die EU-Richtlinien ablösen, muss auch das Medizinproduktegesetz durch ein neues Gesetz abgelöst werden – das Medizinprodukte-Durchführungsgesetz (MPDG).
Folglich muss der Gesetzgeber auch die nationalen Verordnungen anpassen, die die Details zur Umsetzung der Gesetze regeln. Dazu hat er mehrere Möglichkeiten:
- Bestehende nationale Verordnungen durch neue nationale Verordnungen ablösen
- Bestehende nationale Verordnungen ändern
- Bestehende nationale Verordnungen löschen, da die Inhalte der EU-Verordnungen so detailliert sind, dass eine nationale Verordnung nicht mehr notwendig ist
Im Fall der Medizinprodukte-Betreiberverordnung hat sich der Gesetzgeber für die zweite Variante entschieden, im Fall der Medizinprodukte-Sicherheitsplanverordnung für die erste.
Somit wird die MPSV durch die neue Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAMIV) abgelöst.
Diese Änderungen erfolgen durch die MPEUAnpV. Diese ist nicht mit dem MPEUAnpG zu verwechseln. Dieses Gesetz führte u. a. das MPDG ein und änderte das MPG.
Der vollständige Titel der MPAMIV lautet „Verordnung über die Meldung von mutmaßlichen schwerwiegenden Vorkommnissen bei Medizinprodukten sowie zum Informationsaustausch der zuständigen Behörden“.
Sie können sich die MPAMIV als Teil der MPEUAnpV hier herunterladen.Den endgültigen Text finden Sie hier
2. Wen die MPAMIV betrifft
Die MPAMIV betrifft:
- Behörden (BfArM bzw. PEI und Länderbehörden) und Ministerien
- Betreiber von Medizinprodukten (inklusive In-vitro-Diagnostika)
- Patient:innen
- Hersteller von Medizinprodukten (ab Geltungsbeginn der MDR)
- Hersteller von In-vitro-Diagnostika (ab Geltungsbeginn der IVDR)
3. Was die MPAMIV fordert
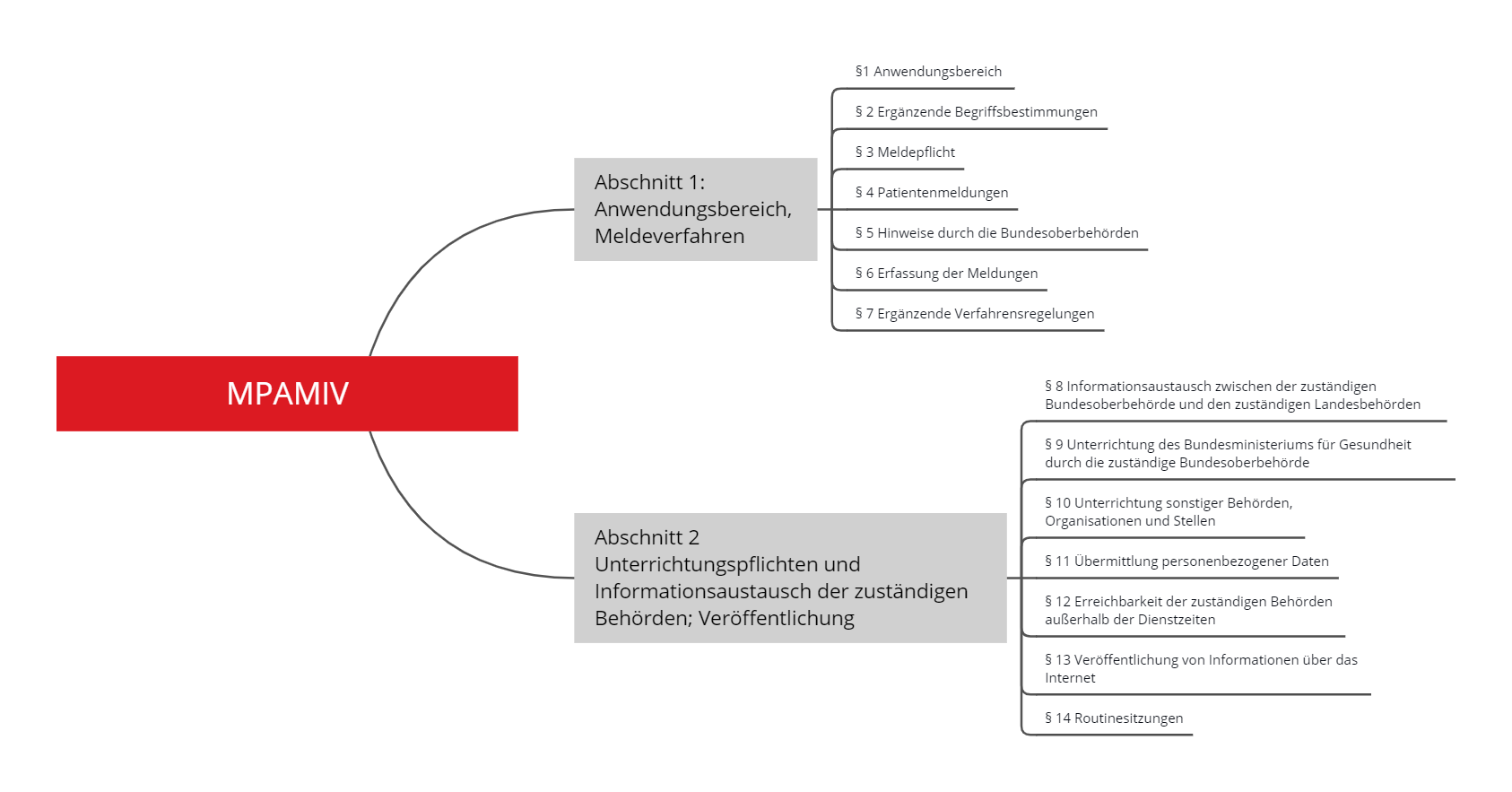
Die Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAMIV) umfasst 13 Paragrafen, die in zwei Abschnitte gegliedert sind.
Abschnitt 1 legt fest, wann und an wen Meldungen über „mutmaßliche schwerwiegende Vorkommnisse“ zu übermitteln sind; Abschnitt 2 regelt die Abstimmung der Behörden untereinander.
a) Anforderungen der MPAMIV an die Betreiber
- „Mutmaßliche schwerwiegende Vorkommnisse“ über Informationssystem (§ 86 MPDG) an die Bundesoberbehörde melden (§ 3)
b) Anforderungen an die Hersteller
- Begründung der zuständigen Bundesbehörde vorlegen (§ 7)
c) Anforderungen an die Behörden und das Bundesministerium für Gesundheit
- Meldungen auf Internetseite veröffentlichen (§ 5)
- Sicherstellung, dass die Meldungen erfasst werden (§6)
- Eingang der Meldungen bestätigen (§ 7)
- Über Handlungsbedarf entscheiden; Maßnahme der Hersteller bewerten (§ 7)
- Meldende Person über Maßnahme des Herstellers und die eigene Risikobewertung informieren (§ 7)
- Informationen untereinander austauschen (§ 8 ff.)
- Erreichbarkeit kommunizieren (§ 12)
- Empfehlungen veröffentlichen (§ 13)
d) Anforderungen an sonstige Personen
- Patient:innen: „Mutmaßliche schwerwiegende Vorkommnisse“ an Arzt oder Bundesoberbehörde melden (§ 4)
4. Was Hersteller jetzt konkret tun sollten
a) Alle regulatorischen Anforderungen studieren
Im Gegensatz zur MPSV enthält die Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAMIV) nur wenige Anforderungen, die die Hersteller betreffen.
Das sollte die Hersteller aber nicht zur Annahme verleiten, dass es weniger Anforderungen gäbe. Diese Anforderungen finden sich lediglich an anderer Stelle!
Relevante regulatorische Anforderungen
Anforderungen an die Vigilanz finden sich u. a. in diesen Dokumenten:
- MDR, z. B. Artikel 87 ff.
- MPDG, v. a. Kapitel 5 (§ 71 ff.)
- Vorgaben der EU bzw. MEDDEV einschließlich
- Manufacturer Incident Report 2020 (PDF-Formular)
- Questions and Answers document regarding the Implementation of the new Manufacturer Incident Report (MIR) Form
- Helptext (Excel-Datei, die angibt, wann welche Informationen verpflichtend sind)
- IMDRF-Codes zu Kategorisierung
b) Prozesse anpassen
Die MPAMIV darf für Hersteller nicht der Auslöser für die Anpassung ihrer Vigilanzprozesse sein. Diesen Auslöser bilden bereits die MDR und das MPDG.
Die Hersteller sollten bei der Überarbeitung ihrer Prozesse u. a. die folgenden Aspekte beachten:
- Der Sicherheitsbeauftragte wird durch die „Verantwortliche Person“ abgelöst. Das bedeutet mehr als ein Austausch der beiden Begriffe in den Verfahrensanweisungen mit „search and replace“. Die Anforderungen und Tätigkeiten sind nicht deckungsgleich.
- Das Zusammenspiel zwischen Post-Market Surveillance und Vigilanz muss ggf. neu geregelt werden.
- Die MDR legt andere Meldefristen fest als die MPSV.
- Hersteller sollten die eigenen Vorgaben bezüglich der Inhalte, der Form und der Adressaten der Meldungen überprüfen.
c) IT-Systeme vorbereiten
Die EU und die nationalen Behörden fordern die Meldungen über („mutmaßliche“) schwerwiegende Vorkommnisse in zunehmendem Maße elektronisch ein.
- Das BfArM stellt(e) dazu PDF-Formulare bereit.
- Zwischenzeitlich werden diese Formulare durch die MIR der EU abgelöst (s. o.).
- Sobald die EUDAMED bereitsteht, müssen die Hersteller die Meldungen dorthin elektronisch übermitteln.
Hersteller sind gut beraten zu prüfen, ob sie die Übermittlung dieser Informationen durch eigene Systeme vereinfachen können. Das ist besonders dann der Fall, wenn sie
- viele Produkte
- in vielen EU-Ländern vermarkten,
- komplexe Zuständigkeiten von Wirtschaftsakteuren haben sowie
- bereits ein System zum Verwalten der Meldungen und Dokumentieren der Maßnahmen betreiben.
5. Kritik an der MPAMIV
a) Unnötige Ausweitung gemäß MDR
Die MPAMIV führt den Begriff „mutmaßliches schwerwiegendes Vorkommnis“ ein:
- den Tod eines Patienten, Anwenders oder einer anderen Person,
- die vorübergehende oder dauerhafte schwerwiegende Verschlechterung des Gesundheitszustands eines Patienten, Anwenders oder anderer Personen,
- eine schwerwiegende Gefahr für die öffentliche Gesundheit.“
Spectaris kritisiert, dass die zusätzliche Definition eines „mutmaßlichen schwerwiegenden Vorkommnisses“ eine Ausdehnung gegenüber dem Begriff des „schwerwiegenden Vorkommnisses“ gemäß MDR bedeutet.
Dieses „bezeichnet ein Vorkommnis, das direkt oder indirekt eine der nachstehenden Folgen hatte, hätte haben können oder haben könnte […]“.
Wie Spectaris anmerkt, ist der Beweis, dass etwas nicht ausgeschlossen werden kann, schwer zu führen und bringt eine Menge „belangloser Meldungen“ mit sich.
b) Unangemessene Meldepflicht
Sowohl Spectaris auch die Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF) kritisieren die Meldepflicht in der jetzigen Form. Beide argumentieren, dass die Ressourcen des BfArMs nicht ausreichen bzw. dass diese Pflicht über die Forderungen der MDR hinausgeht.
Die AOK empfiehlt, die Pflicht, also das „Müssen“, zu ersetzen durch ein „Sollen“.
c) Untersuchung und Abschlussbericht: Unklare Expertise
Die Untersuchungen durch die Bundesbehörden werden generell begrüßt. Die AOK wünscht, dass die Krankenkassen darüber systematisch informiert werden. Die AWMF scheint sich nicht sicher zu sein, ob die Behörden in allen Fällen über die notwendige Expertise dafür verfügen.
d) Weitere Kritik
Es gibt viele Unschärfen, die kritisiert werden, z. B.:
- Fehlende Definition dessen, was ein „beauftragter Sachverständiger“ ist
- Begriff des „Vertreibers“ statt des „Händlers“ bzw. „Wirtschaftsakteurs“
6. Zusammenfassung und Fazit
a) Eine kurze Verordnung
Von der alten Medizinprodukte-Sicherheitsplanverordnung ist wenig geblieben. Die neue Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAMIV) ist mit 14 Paragrafen erstaunlich schlank und enthält wenige Anforderungen an die Hersteller.
b) Anstand ist gefragt
Hersteller sollten auf jeden Fall die Vigilanzanforderungen beachten, die nun in anderen Regularien zu finden sind.
Sie sollten sich auch klar darüber sein, dass ein „Unter-den-Teppich-Kehren“ von Vorkommnissen nicht nur illegal ist, sondern künftig mit höherer Wahrscheinlichkeit auffliegen wird. Zudem fordert er ebenfalls die Patient:innen auf, zu informieren. („Patienten oder deren Angehörige sollen […] den behandelnden Arzt […] oder den Händler, der das Produkt bereitgestellt hat, informieren.“)
Einmal mehr ist anständiges Handeln gefragt: sowohl bei der Entwicklung sicherer Medizinprodukte als auch bei Transparenz und Meldung, falls es doch zu Vorkommnissen kommt.
c) „Gebrauchstauglichkeit“
Die Anforderungen an die Vigilanz waren schon immer über viele Dokumente verteilt. Mussten Hersteller früher das MPG, die MPSV, die MDD und die MEDDEV-Dokumente studieren, sind es jetzt das MPDG, die MPAMIV, die MDR sowie die MEDDEV- und IMDRF-Dokumente.
Damit das nicht zum Problem für die eigenen Mitarbeitenden wird, sollten die Hersteller in den Verfahrensanweisungen klar das Vorgehen im Fall von Vorkommnissen beschreiben, damit sie allen regulatorischen Anforderungen gerecht werden. Deren Abteilungen „Regulatory Affairs“ und „Qualitätsmanagement“ sind jetzt gefordert.
Änderungshistorie
- 2021-08-31: Im Kapitel 3 die Forderungen entsprechend der freigebenden Version der Verordnung aktualisiert. Zuvor bezogen sich die Forderungen auf den Referentenentwurf.
Das Johner Institut unterstützt mit dem Post-Market Radar Hersteller dabei, schlanke und einfach verständliche Systeme für Post-Market Surveillance und Vigilanz aufzusetzen, und übernimmt auf Wunsch diese Prozesse ganz oder teilweise.
Melden Sie sich gleich, z. B. über unsere Kontaktseite.


Guten Morgen Prof. Johner,
vielen Dank für die Erläuterung zum MPAMIV. Gerne würde ich zum Thema „MEDDEV- Dokumente“ (hier erwähnt in der Gebrauchstauglichkeit) eine Frage stellen.
Soweit mir jetzt bekannt, werden in den MEDDEV Dokumenten die EU-Richtlinien als Bezug angezogen. Mir ist bisher nichts bekannt, dass die MEDDEV Dokumente in der EU-MDR angezogen werden können. Können Sie hierzu eine Quelle nennen?
Gibt es nicht Bestrebungen die Leitfäden zur EU-MDR in sogenannten MDCG Dokumenten zu veröffentlichen? Damit wären die MEDDEV Leitfäden hinfällig.
MfG
Sehr geehrter Herr Dittmar,
was Sie beschreiben ist absolut korrekt und entspricht der Planung.
Solange entsprechend Dokumente fehlen wie z.B. bei MEDDEV 2.7/1 ist das Stand der Technik. Die MRI-Forms auf Seiten der MEDDEV adressieren sogar explizit die MDR.
Es gibt also noch etwas Aufräumarbeit zu tun. Wir sind auch unter der MDR die MEDDEV-Dokumente noch nicht ganz los.
Viele Grüße, Christian Johner
Hallo Herr Johner,
bezüglich der Definition eines „mutmaßlich“ schwerwiegenden Vorkommnisse durch die MPAIMV möchte ich gerne noch ergänzen, dass die MDR selbst diesen Begriff in Artikel 87, Absatz 11 ins Spiel bringt (im englischen: Suspected serious incident), den Begriff aber in Artikel 2 nicht definiert. Dies ist national nun geschehen. Aber, ob das im ursprünglichen Sinne der MDR war, möchte ich bezweifeln. Aus meiner Sicht könnte man den Kontext auch so interpretieren, dass die Behörden auf eine Meldung reagieren, bei der noch gar nicht in jedem Fall feststehen muß, dass es sich tatsächlich um ein schwerwiegendes Vorkommnis handelt. Ob man hieraus eine solch umfangreiche Meldepflicht an Anwender und Betreiber ableiten soll, bleibt zweifelhaft.
Bei welchem Vorfall kann denn ein Anwender oder Betreiber denn (sicher) ausschließen, dass es nicht zmindet vorübergehend zu einer schwerwiegenden Beeinträchtigung des Gesundheitszustands hätte kommen können oder kommen könnte. Das bedeutet eine Meldung von so gut wie allen Unzulänglichkeiten, wenn man als Anwender / Bereiber auf der rechtlich sicheren Seite sein will.
Beste Grüße
Gernot Oberländer
Danke Herr Oberländer!
Das ist eine wichtige Ergänzung und Einordnung.
Die MPAMIV folgt mit dieser Linie im Wesentlichen der MPSV. Wenn sich der Eindruck verfestigt, dass die MPAIMV über die MDR hinausgeht, werden Klagen gegen den Gesetzgeber möglich.
Nochmals vielen Dank!
Beste Grüße, Christian Johner
Guten Tag,
mir ist der Meldeweg noch nicht ganz klar. Sofern ich als Händler von einem Kunden eine Information über ein mögliches Vorkommnis erhalte (nicht aktives MP; keine Implantate) muss ich dieses an das BfArM melden oder an den Hersteller, der dann alles weitere in die Wege leitet? Wie verhält es sich, wenn der Hersteller in Europa (also nicht in Deutschland) angesiedelt ist?
Danke für eine kurze Info!
Sehr geehrter Herr Berg,
Händler müssen gemäß Artikel 14 MDR den Hersteller über mutmaßliche Vorkommnisse informieren. Im Falle von importierten Produkten ist auch der Bevollmächtigte und der Importeur zu informieren.
Falls der Händler Grund zur Annahme hat, dass von dem Produkt eine schwerwiegende Gefahr ausgeht, muss dies gemäß §81 MPDG dem BfArM über das DMIDS gemeldet werden.
Freundliche Grüße
Luca Salvatore
Guten Tag,
vielen Dank für die Erläuterungen!
Wie ist den ein „mutmaßliches Vorkommnis“ definiert, über welche Händler und Importeure informieren müssen?
In der MDR ist nur „Vokommnis“ und „schwerwiegendes Vorkommnis“ definiert und in der MPAMIV ist nur „mutmaßlich schwerwiegendes Vorkommnis“ definiert.
MfG
Carina Gasch
Guten Tag Frau Gasch,
der korrekte Begriff in der MDR lautet „schwerwiegende Gefahr“ bzw. in der englischen Fassung „serious risk“.Der Begriff ist leider nicht definiert. In MDCG 2023-3 unter 18. finden Sie aber eine Interpretation.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
Vielen Dank für die Referenz.
In Artikel 13 (8) und 14 (5) der MDR wird ja aber eben auch von „mutmaßlichen Vorkommnissen“ gesprochen. Ist dieser Begriff nirgends definiert? Wie würden Sie denn Begriff definieren?
Vielen Dank im Voraus?
MfG Carina Gasch
Genau. Die MDR spricht auch von „mutmaßlichen Vorkommnissen“ im Zusammenhang mit der Meldung an den Hersteller bzw. Importeur (nicht die Meldung an die Behörde).
Damit sind Beschwerden gemeint, von denen der Händler/Importeur erfährt und die einem Vorkommnis (gemäß Definition in Artikel 2 64. MDR) entsprechen. Dabei ist es unerheblich, ob es sich tatsächlich um ein Vorkommnis handelt oder die Beschwerde vielleicht nicht berechtigt war, z.B. da das Produkt spezifikationsgemäß funktioniert hat. Denn dies soll der Hersteller im Rahmen seines Reklamationsprozesses selbst bewerten bzw. untersuchen. Deshalb spricht die MDR hier von „mutmaßlich“.
Freundliche Grüße
Luca Salvatore
Guten Tag,
ich hätte eine Frage zu folgendem Absatz im Artikel oben:
„b) Anforderungen an die Hersteller
Fotodokumentation des Produkts vor dessen unvermeidbarer Zerstörung erstellen (§ 7)“
Im §7 der MPAMIV finde ich diese Forderung nicht. Steht es in einem anderen §?
Wo ich diese Forderung gefunden habe ist im §72 Absatz (6) des MPDG.
Vielen Dank im Voraus
Beste Grüße
Nadine Langguth
Großartige Frage, Frau Langguth!
Die Forderung nach der Fotodokumentation war im Entwurf der Verordnung enthalten, in der endgültigen Version nicht mehr. Wir werden den ganzen Artikel nochmals durchgehen, ob noch an anderen Stellen Inhalte so geändert wurden.
Danke für Ihren so wertvollen Hinweis!
Viele Grüße, Christian Johner
Nun wäre es nich interessant zu wissen was die MDR unter „Schwerwiegender Gefahr“ versteht, damit ich den Meldepflichten auch nachkommen kann!?
danke für ein kurzes Statement dazu
Sehr geehrter Herr Leber,
in der Tat definiert die MDR oder das MPDG den Begriff „schwerwiegende Gefahr“ nicht. Als Hilfestellung/Orientierung könnte lediglich der Begriff „schwerwiegende Gefahr für die öffentliche Gesundheit“ (Artikel 2 Punkt 66) herangezogen werden.
Viele Grüße
Guten Tag,
da ich mich jetzt schon seit etlicher Zeit durch die Gesetzgebung mühe und bis jetzt noch auf keinen grünen Zweig gekommen bin, stelle ich meine Frage nun an Sie:
Findet sich in der aktuellen Gesetzgebung irgendwo die explizite Meldepflicht für einen Rückruf? Oder wird dieser als „Maßnahme“ betrachtet und fällt dann unter Sicherheitskorrekturmaßnahmen im Feld?
Für eine kurze Rückmeldung bin ich sehr dankbar und verbleibe mit freundlichen Grüßen
Beate Jenne
Liebe Frau Jenne,
Sie meinen die Meldepflicht für den Medizinproduktehersteller bei Rückrufen?
Das geltende Recht in den Mitgliedstaaten der EU wird durch die Medizinprodukteverordnung MDR 2017/745 vorgegeben.
Dort heißt es in Artikel 87:
Da sich Sicherheitskorrekturmaßnahmen („FSCA“) nach MDR Artikel 2 (68) definitionsgemäß auf „schwerwiegende Vorkommnisse“ beziehen, wären nach europäischem Recht nur die damit verbundenen Rückrufe meldepflichtig, also Rückrufe zur Verhinderung von schwerwiegenden (!) Vorkommnissen.
Der Begriff Rückruf hat nach der MDR Artikel 2 (62) keinen Risikobezug. Denn es heißt einfach:
Demnach sind nicht alle Rückrufe meldepflichtig!
Beschäftigen Sie sich gerne auch mit diesem Dokument der EU (Guidance MEDDEVs):
https://ec.europa.eu/health/sites/default/files/md_sector/docs/md_guidance_meddevs.pdf
Dort sind unter Abschnitt „2.12 Post-Market surveillance“ die geltenden Vorgaben zum Meldewesen bei Medizinprodukten aufgeführt.
Ich hoffe, damit habe ich Ihnen langwierige Recherchen erspart…
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Prof. Johner,
zur Einordnung des neuen Begriffs des „mutmaßlichen schwerwiegenden Vorkommnisses“ würde ich gerne die folgende Deutung in den Raum stellen: In Artikel 87 Absatz 3 fordert die MDR, schwerwiegende Vorkommnisse zu melden, nachdem ein Kausalzusammenhang oder ein „durchaus möglicher Kausalzusammenhang“ festgestellt wurde. Ich würde es für denkbar halten, dass mit dem neuen Begriff einfach dieser Bezug zu Artikel 87 Absatz 3 leichter beschrieben werden können soll. Außerdem wird in Absatz 7 ja gefordert, dass ein Hersteller auch dann eine Meldung abgeben soll, wenn er sich unsicher ist, ob das Vorkommnis zu melden ist. Insofern würde ich in der Forderung der MPAMIV keine Verschärfung gegenüber der MDR erkennen – wenn denn diese Deutung richtig ist.
Wie sehen Sie das?
Schöne Grüße
Karsten Roeseler
Sehr geehrter Herr Roeseler,
danke für Ihre wertvollen Gedanken! Ich teile Ihre Meinung, dass man die Definition auch in Ihrem Sinn verstehen kann und nicht als Verschärfung, wie das Spectaris formuliert.
Diese Diskussionen sind einmal mehr unvollständigen und manchmal inkonsistenten Definitionen geschuldet.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
vielleicht können Sie bei Klärung des Schicksals der früheren Vorschrift, vor zerstörender Prüfung des Medizinprodukts eine Oberbehörde anzusprechen (§ 12 Abs. 2 S. 3 MPSV a.F.), helfen. Ich habe den Eindruck, dass ein solches Gebot heute nicht länger besteht. Ist das richtig oder habe ich etwas übersehen?
Mit Dank und freundlichen Grüßen
Ulrich Foerste
Lieber Herr Kollege Foerste,
danke für Ihre wichtige Frage!
Sie finden eine vergleichbare Anforderung im §72 des MPDG.
Mit den besten Grüßen, Christian Johner
Guten Tag,
Ich habe eine Frage zu Abschnitt 2. Wenn der Hersteller die Meldung an das BfarM geschickt hat, wird diese Meldung auch an andere EU-Behörden weitergeleitet? Oder muss der Hersteller jedes Land einzeln benachrichtigen?
Für eine kurze Rückmeldung bin ich sehr dankbar und verbleibe mit freundlichen Grüßen
William Trümmer
Sehr geehrter Herr Trümmer,
derzeit ist die EUDAMED noch nicht voll funktionsfähig. Daher müssen Sie die Meldungen in den einzelnen Ländern leider noch einzeln machen.
Danke für die wichtige Frage!
Viele Grüße
Christian Johner
Guten Tag,
ich sehe in der MPAMIV keine klare Angabe mehr (anders als in der MPSV) zur Meldung von Cybersicherheits relevanten Meldungen.
Das BfArM selbst hat damals erwähnt, „…dass die in der Medizinprodukte-Sicherheitsplanverordnung (MPSV) genannten Meldekriterien und -pflichten auch für Risiken bzw. Ereignisse gem. § 2 Abs. 1 MPSV gelten, die im Zusammenhang mit der Vernetzung von Medizinprodukten stehen“
Soll dies in Zukunft alles über, das BSIG, BSI-KritisV, KRITIS-DachG, NIS-2-Richtlinie bzw. der deutschen Umsetzung dieser laufen?
Mir scheint, dass sich die Regelungen auf zu viele Gesetze und Verordnungen verteilen, um eine wirksame Regelung zu generieren.
Was halten Sie davon oder übersehe ich hier einen entscheidenen Punkt.
Lieber Herr Dr. Breß,
es ist, wie Sie schreiben: Die MPAIMV enthält im Gegensatz zur MPSV diesen Passus nicht.
Dennoch sind „schwerwiegende Vorkommnisse“ zu melden, auch wenn sie auf Probleme mit der Cybersicherheit zurückzuführen sind. D.h. ein Verzicht auf diese Meldung in der Annahme, man habe über andere Kanäle bzw. an andere Behörden gemeldet, ist ein Irrtum und Rechtsbruch.
Ich teile Ihre Meinung, dass hier unter Umständen Doppelarbeit zu leisten ist. Allerdings sind die Zielsetzungen und Bewertungen anders:
Bei den Meldungen nach MDR bzw. MPAMIV ist das Bewertungskriterium der Schweregrad möglicher Schäden für Patienten, Anwender und Dritter. Bei den Meldungen z.B. gemäß KRITIS ist das Bewertungskriterium das Aufrechterhalten der Versorgungssicherheit.
Was Sie ansprechen, ist das Dilemma von orthogonalen Regulierungen.
Danke für Ihre wichtigen Gedanken!
Viele Grüße
Christian Johner