
Die dritte Ausgabe der ISO 14971 steht seit Dezember 2019 bereit.
Hersteller sollten sich mit den neuen und geänderten Anforderungen dieser Norm vertraut machen. Noch im Dezember 2019 hat die FDA die 3. Ausgabe Norm der ISO 14971 anerkannt. Mehr zu den Übergangsfristen weiter unten.
1. Dritte Ausgabe der ISO 14971
Die dritte Ausgabe („third edition“) der ISO 14971 folgt auf die Vorgängernorm, die ISO 14971:2007 („second edition“).
Parallel hat die ISO auch die ISO 24971 überarbeitet, die ebenfalls als Entwurf vorliegt. Diese „Erläuterungsnorm“ gewinnt an Bedeutung, weil sie nun einige der nicht-normativen Anhänge der alten ISO 14971 enthält.
2. Die Änderungen im Überblick
a) Neue Kapitelstruktur
Als erstes fällt die neue Kapitelstruktur auf. Die ISO 14971:2019 folgt nun dem üblichen Aufbau, der mit den folgenden Kapiteln beginnt:
- Anwendungsbereich („Scope“)
- Normative Verweise („Normative Reference“)
- Begriffsdefinitionen („Terms and definitions“)
Durch das neue Kapitel mit den normativen Verweisen verschiebt sich die Nummerierung. Die ISO 14971:2019 hat nun zehn Kapitel.
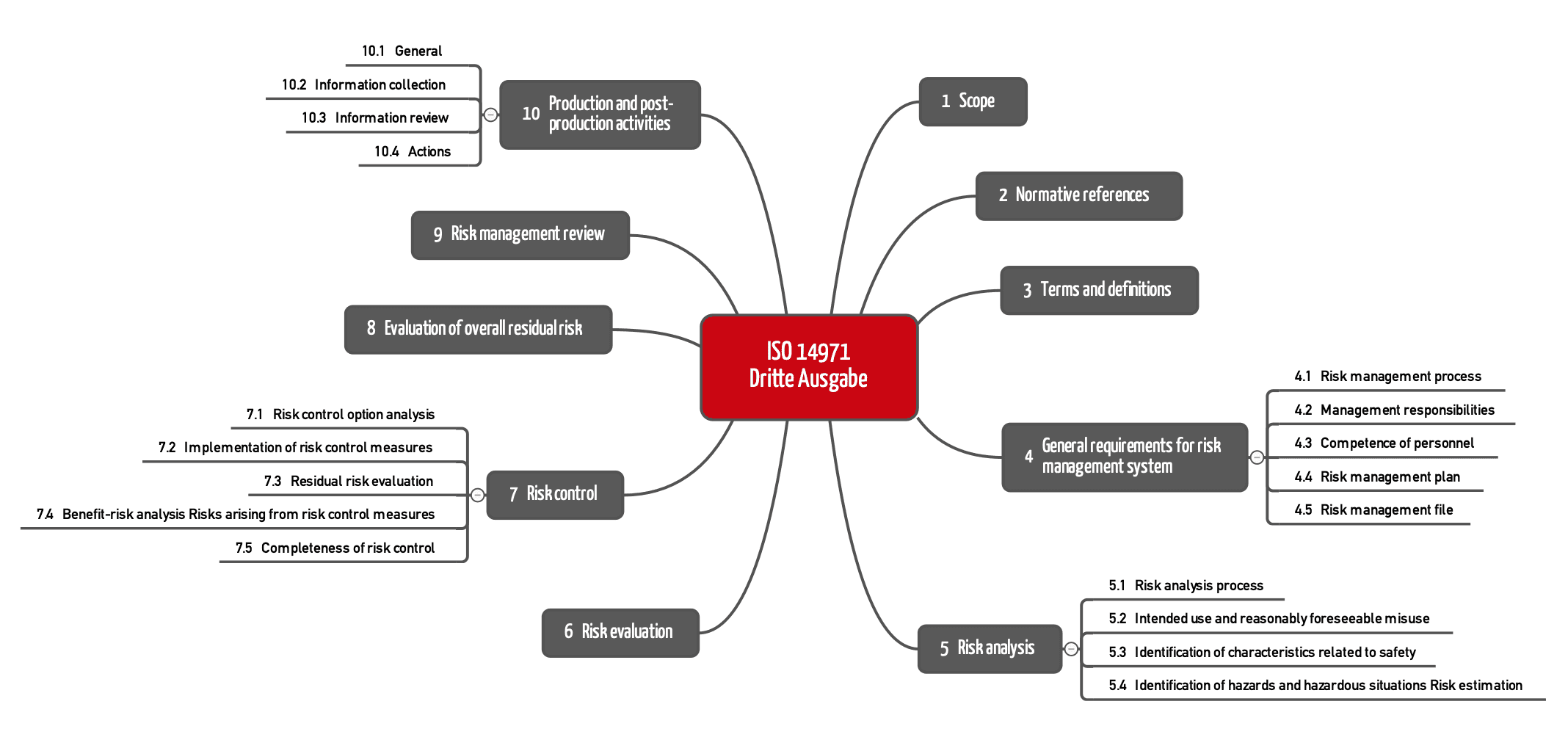
Bereits die Kapitelstruktur offenbart einen weiteren Unterschied: Die Anforderungen an die nachgelagerte Phase sind umfangreicher und in vier Unterkapitel (10.1 bis 10.4) unterteilt.
b) Höhere Relevanz des Nutzen-Risiko-Verhältnisses?
Die ISO 14971:2019 behauptet, noch deutlicher Wert auf den Nachweis zu legen, dass der Nutzen die Risiken überwiegt. Sie ergänzt die fehlende Definition des Begriffs Nutzen („Benefit“).
Beispiele für diesen Nutzen sind:
- Schnellere Genesung, vollständigere Genesung
- Heilung mit weniger Nebenwirkungen
- Genauere Diagnose
- Bessere öffentliche Gesundheitsversorgung
Damit ist klar, dass mit Nutzen der medizinische Nutzen gemeint ist und nicht etwa ein höherer ökonomischer Nutzen für den Betreiber.
Wirklich neue Anforderungen stellt die Norm nicht. Unverändert fordert sie, dass es die Aufgabe des Managements ist, die Risikopolitik festzulegen. Diese muss sich am Stand der Technik orientieren. Zumindest eine Definition des Begriffs „Stand der Technik“ ergänzt die dritte Ausgabe der ISO 14971:2019.
Dieser Stand der Technik ist nicht mit dem Stand der Wissenschaft zu vergleichen. Er entspricht eher allgemein anerkannten technischen und medizinischen „Good Practices“.
Neu ist in der dritten Ausgabe der ISO 14971, dass die Hersteller für die Bewertung der Einzelrisiken andere Akzeptanzkriterien festlegen können als für die Bewertung des gesamten Restrisikos. Die Akzeptanzkriterien für die Einzelrisiken können verwendet werden, um über die Notwendigkeit von Maßnahmen zur Risikobeherrschung zu entscheiden. Die Akzeptanzkriterien für das Gesamtrisiko können herangezogen werden um zu entscheiden, ob das Produkt in den Verkehr gebracht werden darf.
Die Norm verpflichtet die Hersteller dazu, die Methoden zu beschreiben, mit denen die Akzeptanz des Gesamtrestrisikos bestimmt wird.
c) IT Security im Scope
Die dritte Ausgabe der ISO 14971 nimmt Risiken durch mangelnde „data and system security“ explizit in den Scope mit auf. Sie stellt dafür aber keine spezifischen Anforderungen.
Besonders im deutschsprachigen Raum besteht die Gefahr, dass die Hersteller die Schutzziele „Safety“ und „Security“ nicht präzise unterscheiden, weil beide mit „Sicherheit“ übersetzt werden.
Während die Abwägung von medizinischem Nutzen und „Safety-Risiken“ Sinn ergibt, kann die Abwägung von medizinischem Nutzen und „Security-Risiken“ in die Irre führen. Eine Erhöhung der Security kann sogar zu Lasten der Safety gehen.
Überlassen Sie die Sicherheit Ihrer Patienten nicht dem Zufall
Gehen Sie mit einem Pentest des Johner Instituts auf Nummer sicher!
d) Vernünftigerweise vorhersehbarer Fehlgebrauch ist zu berücksichtigen
Die ISO 14971:2019 ergänzt die explizite Forderung, den vernünftigerweise vorhersehbaren Fehlgebrauch zu analysieren. Sie definiert diesen „reasonably forseeable misuse“ wie folgt:
Dieser Fehlgebrauch kann absichtlich oder unabsichtlich erfolgen. Ein Beispiel wäre die Nutzung eines Produkts, ohne vorher die Gebrauchsanweisung studiert zu haben.
Beachten Sie, dass die deutsche Übersetzung von „misuse“ nicht Missbrauch, sondern Fehlgebrauch ist. Der Begriff „Missbrauch“ assoziiert etwas Böswilliges, was beim „misuse“ aber gar nicht vorliegen muss. Auch eine gut gemeinte „Fehlanwendung“ ist zu berücksichtigen.
e) Sicherheitsbezogene Charakteristiken sind zu identifizieren
Neu ist zwar das Kapitel zu den sicherheitsbezogenen Charakteristiken, aber die Anforderungen sind es nicht. Diese Charakteristiken des Produkts müssen die Hersteller qualitativ und quantitativ erfassen – am besten mit Angabe von Grenzwerten, die für die Sicherheit des Produkts wesentlich sind. Alle Kenner der IEC 60601-1 denken sofort an die wesentlichen Leistungsmerkmale. Zu Recht.
Das Johner Institut empfiehlt, insbesondere die Systemanforderungen daraufhin zu untersuchen, ob ein Risiko folgen könnte, wenn diese nicht oder nicht im spezifizierten Maß erfüllt sind.
f) Anforderungen an die Produktion und die nachgelagerte Phase
Die auffälligste Änderung betrifft das Risikomanagement in der Produktion sowie in der der Produktion nachgelagerten Phase – sprich: der Post-Market-Phase. Die Anforderungen erinnern stark an die der MDR.
Sowohl die MDR als auch die dritte Ausgabe der ISO 14971 fordern ein proaktives Sammeln und Bewerten der Daten aus den Phasen nach der Entwicklung. Die MDR spricht von einem Prozess, die ISO 14971 von einem System.
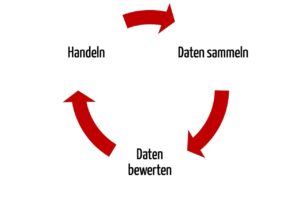
Ähnlich wie die MDR legt auch die Norm die Informationsquellen fest, die auf jeden Fall betrachtet werden müssen. Darunter finden sich z.B. öffentliche Informationen, Informationen zum Stand der Technik und Informationen, die bei der Installation, Anwendung und Wartung des Produkts entstehen.
Die Informationen müssen daraufhin bewertet werden, ob
- neue, bisher nicht betrachtete Gefährdungen zu beachten sind,
- die Risiken (Wahrscheinlichkeiten und Schweregrade von Schäden) richtig bewertet sind und
- die Risiken weiterhin akzeptabel sind, z.B. weil sich der Stand der Technik geändert hat.
Abhängig von den Ergebnissen dieser Bewertung muss der Hersteller handeln. Konkret nennt die dritte Ausgabe der ISO 14971 Handlungen, die das Medizinprodukt betreffen (z.B. neue risikominimierende Maßnahmen implementieren) und Handlungen, die das Risikomanagement (z.B. den Risikomanagementprozess) betreffen.
Lesen Sie hier mehr zum Thema nachgelagerte Phase und Post-Market Surveillance.
3. Rechtliche Verbindlichkeit
a) Europa
Die EN ISO 14971:2019 ist inzwischen sowohl für MDR als auch IVDR harmonisiert. Die Durchführungsbeschlüsse zur Harmonisierung sind für die MDR und IVDR online zu finden.
b) USA / FDA
Die FDA wird die zweite Ausgabe der Norm aus dem Jahr 2007 noch bis Ende 2022 gestatten. Spätestens danach besteht die FDA auf der Anwendung der dritten Ausgabe der ISO 14971.
4. Fazit
Die dritte Ausgabe der ISO 14971 ist noch besser als die bereits gute zweite Ausgabe. Viele Änderungen sind redaktioneller Art und sorgen für mehr Klarheit und Stringenz.
Besonders erwähnenswert sind die präzisierten Anforderungen an die nachgelagerte Phase. Dennoch bleibt der Umfang der Änderungen beschränkt, sodass „Version 2.1“ der Sache eher gerecht geworden wäre. Bedauerlich ist insbesondere:
- Viele hilfreiche Anhänge sind in die ISO 24971 verschoben worden. Das macht die ISO 14971 zwar schlanker, zwingt aber die Hersteller, eine zweite Norm zu kaufen.
- Einige Erläuterungen sind ganz verschwunden. So hatte die alte ISO 14971 noch klargestellt, dass sich das Risiko eben nicht aus der Multiplikation von Schweregrad und Wahrscheinlichkeit von Schäden berechnet. Wie kann eine so zentrale und berechtigte Feststellung verschwinden angesichts der Tatsache, dass 95 % der Hersteller genau dies tun?
- Es ist anzunehmen, dass die EU die Forderungen der MDR zum Risikomanagement durch die dritte Ausgabe nur teilweise abgedeckt sieht. Dadurch drohen wieder zusätzliche Anforderungen und normative Interpretationen in den Z-Anhängen.
- Das Zusammenspiel des Risikomanagements mit der klinischen Bewertung beschreibt die dritte Ausgabe der ISO 14971 gar nicht, die überarbeitete ISO 24971 nur rudimentär.
- Es ist nachvollziehbar, dass das Normengremium die Norm an die sonst übliche Kapitelstruktur angleichen wollte. Diese redaktionelle Änderung wird dazu führen, dass die meisten Hersteller ihre Vorgabedokumente (Verfahrensanweisungen, Arbeitsanweisungen, Templates) usw. daraufhin untersuchen müssen, ob die Referenzen auf die Kapitelstruktur noch stimmen. Viel Arbeit, die nicht der Patientensicherheit zugute kommt.
Trotz dieser Wehmutstropfen dürften die Hersteller mit dieser dritten Ausgabe der ISO 14971 gut leben können. Weniger ist eben manchmal mehr.
5. Aktuelles
Seit Frühjahr 2022 ist auch die deutsche Version DIN EN ISO 14971:2022-04 veröffentlicht. Diese beinhaltet die harmonisierte Fassung EN ISO 14971:2019 sowie den Anhang A11:2021, der in erster Linie noch einmal klarstellt, dass die Norm mit MDR und IVDR vereinbar ist.
Änderungshistorie
- 2022-08-25: Hinweis auf deutsche Fassung DIN EN ISO 14971:2022-04 sowie Harmonisierung ergänzt
- 2021-02-23: Hinweis ergänzt, dass „misuse“ mit „Fehlgebrauch“ oder „Fehlanwendung“ und nicht mit „Missbrauch“ übersetzt werden sollte. Es geht auch um „gut gemeinte Fehlanwendungen“.
- 2020-11-16: Hinweis auf Standardization Request und damit auf die Planung einer Harmonisierung der EN ISO 14971:2019 eingefügt

Hallo,
zum noch besseren Verständnis würde ich folgendes ergänzen/umformulieren, nachdem ich es mir selbst noch einmal durchgedacht habe:
So hatte die alte ISO 14971 noch klargestellt, dass sich das Risiko eben nicht aus der *Multiplikation* von Schweregrad und Wahrscheinlichkeit von Schäden als eine Risikozahl berechnet, sondern dass für jede Kombination von Schweregrad und Wahrscheinlichkeit eine Einstufung/Bewertung (sinnvollerweise in einer 2D Matrix) zu treffen ist.
So kann daraus folgend eine Kombination aus 2 (Auftreten sehr selten) und 5 (Schweregrad katastrophal) als nicht akzeptables Risiko eingestuft werden, aber eine Kombination aus 5 (Auftreten häufig) und 2 (Schweregrad geringfügig) als akzeptables Risiko.
Bei einer reinen Bewertung des Produkts dieser beiden „Faktoren“ wäre die Einstufung gleich.
beste Grüße aus Dinkelsbühl
Hallo,
mir ist ein Punkt aufgefallen der typisch für das mittlerweile destruktive Vorgehen der Regularien und Normen gegenüber kleinen und innovativen Firmen ist.
Sie schreiben zum vernünftigerweise vorhersehbaren Missbrauch: „Ein Beispiel wäre die Nutzung eines Produkts, ohne vorher die Gebrauchsanweisung studiert zu haben.“
… das Problem ist also der Anwender, nicht das Produkt. Dennoch muss das Produkt dem entgegenwirken.
Ist es nicht grob fahrlässig vom Anwender ein Medizinprodukt zu verwenden, ohne die GA gelesen zu haben?
Wie soll so etwas verhindert werden? Es ist ja davon auszugehen, dass der Anwender auch Warnzeichen ignoriert, wenn er schon die Anleitung nicht ließt.
Danke für Ihre hervorragende Webside, Sie haben mir damit schon oft geholfen.
Gruß Michael Balling
Sehr geehrter Herr Balling,
danke für Ihre wertschätzende Rückmeldung!
Ich teile Ihre Meinung, dass viele Gesetze und Normen für kleine Firma zur Herausforderung geworden sind.
Gleichzeitig ist es unser aller Ziel, jederzeit sicherzustellen, dass der Nutzen durch die Produkte die Schäden bzw. Risiken überwiegt. Dass Anwender die Gebrauchsanweisung nicht oder nicht richtig lesen, zählt zum vorhersehbaren Missbrauch. Den müssen die Hersteller berücksichtigen. Dabei können Sie aber die Wahrscheinlichkeit mit berücksichtigen, dass es diesen Missbrauch gibt.
Auch Warnzeichen bilden, wie Sie richtig ausführen, keine sehr hohe Sicherheit. Daher verlangen die Regularien, auch die ISO 14971 zuerst inhärente Sicherheit anstreben, wenn das nicht möglich ist, Schutzmaßnahmen ergreifen, und erst als letzte Möglichkeit Informationen wie Warnungen und Gebrauchsanweisungen als Mittel zur Risikobeherrschung nutzen.
Mit den besten Grüßen
Christian Johner
Sehr geehrter Professor Johner,
Sie schreiben in Abschnitt 1 oben:
„Noch ist unklar, ob die EU-Kommission für die Medizinprodukteverordnung (MDR) die neue Version harmonisiert, die dann möglicherweise als EN ISO 14971:2020 geführt wird.“
Nun wurde von der EU-Kommission Ende Oktober (29.10.2020) ein neuer Entwurf des „standardization requests“ unter MDR/IVDR an die CEN/Cenelec gesendet. Dort ist auch die EN ISO 14971:2019 gelistet!
Ist dementsprechend zu hoffen, dass diese EN ISO 14971:2019 doch unter der MDR harmonisiert werden soll?
Mit freundlichen Grüßen
Boetcher
Sehr geehrter Herr Boetcher,
genauso ist es! Ich hatte den Beitrag noch nicht überarbeitet. Danke für den wichtigen Hinweis. Dank Ihrer Hilfe konnte ich direkt auf den Standardization Request hinweisen.
Im morgigen Instituts-Journal habe ich ebenfalls den Link auf diesen Standardisation Request eingefügt.
D.h. die Hoffnung auf eine Harmonisierung ist da, die Planung mit einer Harmonisierung bis 2024 ist aber eher als etwas optimistisch zu sehen.
Nochmals vielen Dank!
Beste Grüße, Christian Johner
Hallo Herr Johner,
„misuse“ wird leider häufig mit „Missbrauch“ übersetzt.
Eine bessere Übersetzung wäre jedoch z.B. „Fehlgebrauch“. Die 14971-2019 übersetzt „misuse“ mittlerweile mit „Fehlanwendung“.
Bei unseren Projekten hat es sich auch gezeigt, dass der „Missbrauch“ wirklich nur einen kleinen Anteil bei der Analyse zum Thema „Fehlgebrauch“ einnimmt.
Ich habe das Gefühl, allein diese unglückliche Übersetzung hat bei vielen deutschen Firmen zu unnötigen Mißverständnissen und Fehlleitungen geführt…
Viele Grüße aus Freiburg,
Oguz Özcelik
Sehr geehrter Herr Özcelik,
danke für Ihren sehr wichtigen Hinweis! Ich stimme allen Ihren Überlegungen zu.
Ich werden den Beitrag gemäß Ihrer Gedanken optimieren und genau auf diese Thematik hinweisen.
Besten Dank und viele Grüße, Christian Johner
Hallo liebes Johner Team,
evtl. arbeiten Sie bereits daran, aber ich denke, dass es sinnvoll wäre den Artikel um die DIN EN ISO 14971:2022 bzw. eben die EN ISO 14971:2019 + A11:2021 zu ergänzen und was dies für die wohl anstehende Harmonisierung bedeutet.
Viele Grüße und danke für Ihre vielen guten Informationen,
Siegfried Bolek
Vielen Dank für den Hinweis, Herr Dr. Bolek! Ich hatte geplant, die Harmonisierung der neuen Version im Beitrag einzufügen, sobald die Harmonisierung offiziell verkündet wird. Leider hat das in der letzten Publikation der harmonisierten Normen noch nicht geklappt, aber wir warten täglich darauf…
Fest steht, dass die EN ISO 14971:2019 + A11:2021 bereits harmonisierungsfähig ist. Sie fehlt halt nur noch offiziell im Amtsblatt der EU als harmonisierte Norm.
Herzliche Grüße
Christian Rosenzweig
Sehr geehrter Herr Prof. Johner,
muss eine Auftragsfertiger (Übernahme der des gesamten Herstellungsprozesses im Zuge der Auftragsfertigung) für das im Auftrag gefertigte Produkt eine Risikomangementakte führen?
Herzlichen Dank vorab für Ihre Antwort!
LG Astrid Unterkreuter
Liebe Frau Unterkreuter,
das ist eine spannende Frage und wird oft falsch interpretiert. Die Verpflichtung zum Risikomanagement nach ISO 14971 kommt aus der EU-MDR und richtet sich an den Hersteller eines Medizinproduktes. Wenn dieser Teile seiner Wertschöpfung (Teile der Entwicklung und/oder Fertigung oder beides gesamt) an einen Dienstleister auslagert, bleibt die Verantwortung dennoch beim Hersteller. Er kann aber die operative Durchführung des Risikomanagements – zumindest in Teilen – an einen Lieferanten per QSV (Qualitätssicherungsvereinbarung) delegieren (z.B. Risikoanalysen nach IEC 60812 mit FMEA-Methoden). Letztendlich macht der Medizinproduktehersteller aber das Risikomanagement in der der Produktion nachgelagerten Phase des Produktlebenszyklus. Deshalb ist es sinnvoll, dass der Medizinproduktehersteller auch detailliert die Risikomanagementakte nach ISO 14971 kennt und selbst pflegt.
Beantwortet das Ihre Frage ausreichend?
Herzliche Grüße
Christian Rosenzweig
Hallo liebes Johner Team,
die MDR fordert in Anhang II unter 5. „Informationen über die Nutzen-Risiko-Analyse gemäß Anhang I Abschnitt 1 und 8“ als Teil der technischen Dokumentation. Abgesehen davon, dass in den Abschnitten keine Nutzen-Risiko-Analyse gefordert wird, sondern nur, dass die Risiken vertretbar im Verhältnis zum Nutzen sein sollen, frage ich mich welche Informationen hier genau gefordert werden? Ist damit die Beurteilung der Vertretbarkeit des Nutzen-Risiko-Verhältnisses gemeint, wie sie auch in Artikel 61 (1) als Teil der klinischen Bewertung beschrieben wird?
In der ISO 14971 ist in Kapitel 7.4 ebenfalls von einer Nutzen-Risiko-Analyse die Rede, wobei es sich dabei wohl nicht um die in der MDR geforderte handeln kann, da sie nur optional anzuwenden ist, wenn ein Restrisiko nicht akzeptabel ist aber auch nicht weiter vermindert werden kann.
In der Norm wird stattdessen von „Gesamt-Restrisiko im Verhältnis zum Nutzen“ (Kapitel 10) gesprochen, was wohl dem entspricht was in der MDR wiederum als Nutzen-Risiko-Abwägung definiert ist und im Endeffekt in Anhang II unter 5. als Teil der technischen Dokumentation gefordert wird.
Beste Grüße
Joscha Hüttel
Lieber Herr Hüttel,
vielen Dank für Ihren Kommentar, der bestimmt auch anderen Lesern dieser Seite eine Hilfe ist. Tatsächlich darf man den einzelnen Worten im Anhang II, Abschnitt 5 nicht zu viel Bedeutung beimessen. Die Benannten Stellen verstehen unter diesem Punkt die Darstellung des gesamten Risikomanagement-Prozesses (Verfahrensanweisung und/oder Plan) und der entsprechenden Nachweisdokumente zur Umsetzung. Also letztlich die gesamte Risikomanagement-Akte! So haben wir es in der Praxis erlebt und so stellt es auch das Positionspapier des Team-NB dar (ab Seite 17).
Zum Begriff „Nutzen-Risiko-Abschätzung“ noch eine Anmerkung: Wie Sie schon schreiben, ist das die Betrachtung der Gesamtrestrisiken im Verhältnis zum Nutzen des Produktes. Diese Betrachtung wird im Risikomanagement und im Bericht zur klinischen Bewertung aufgeführt und stellt sozusagen die Quintessenz des Risikomanagements dar.
In Kapitel 7.4 der ISO 14971:2019 wird der Begriff „Nutzen-Risiko-Analyse“ ebenfalls verwendet, meint dort aber nur das nach Maßnahmen weiterhin „unvertretbare“ Einzelrestrisiko, das Sie durch eine entsprechende Nutzen-Risiko-Abschätzung doch noch „vertretbar“ machen können. Das wird leider häufig verwechselt.
Fazit: Legen Sie in der Technischen Dokumentation Ihre gesamte Risikomanagement-Akte mit allen entsprechenden Inhalten ab, damit Sie Abschnitt 5 des Anhang II der MDR erfüllen.
Herzliche Grüße
Christian Rosenzweig
Hallo liebes Johner-Team,
ich habe ein möglicherweise, banale Frage zu risikomindernden Maßnahmen. Wie könnte eine solche aussehen, dass nach Anwendung dieser nicht nur die Auftretenswahrscheinlichkeit, sondern auch auch der Schweregrad verringert wird? Beide Möglichkeiten werden in der 14971 erwähnt, jedoch habe ich bislang bei den Unternehmen, bei denen ich bislang gearbeitet habe, letztere nie in einer Risikomanagementakte gesehen.
Vielen Dank und beste Grüße
Thomas Hertwig
Lieber Thomas,
danke für Deine Frage! Tatsächlich zielen Risikobeherrschungsmaßnahmen fast immer auf die Wahrscheinlichkeit eines Gesundheitsschadens. Ein Beispiel für die Absenkung des Schweregrades wäre: Ein mit Netzspannung betriebenes Medizinprodukt kann einen tödlichen Stromschlag hervorrufen. Wenn die Maßnahme darin besteht, das Produkt stattdessen nur mit Standard-Batterien mit niedriger Spannung zu betreiben, dann ist zwar ein Stromschlag aufgrund der gleichen Ursachen wie zuvor auch noch möglich, aber der Schaden wäre vermutlich deutlich geringer und eher am anderen Ende der Schweregrad-Achse.
Schöne Grüße nach Bad Homburg
Christian
Lieber Christian,
danke für das anschauliche Beispiel.
Das hilft sehr.
Viele Grüße
Thomas