
Unter „Design Input“ versteht man die Entwicklungsvorgaben, an die nicht nur die FDA konkrete Forderungen stellt.
| Inhaltsübersicht |
|---|
| Definition „Design Input“ » |
| Regulatorische Anorderungen » |
| Inhalte des Design Inputs » |
| Risikoanalyse als Design Input » |
Definition des Begriffs „Design Input“
Die FDA definiert den Begriff in 21 CFR 820.3(f) wie folgt:
„Design input means the physical and performance requirements of a device that are used as a basis for device design.“
Auch die ISO 13485 nutzt diesen Begriff, allerdings ohne ihn zu definieren.
Regulatorische Anforderungen
a) FDA 21 CFR 820.30
Die FDA legt auch die Anforderungen an den Design Input fest. Sie schreibt:
Each manufacturer shall establish and maintain procedures to ensure that the design requirements relating to a device are appropriate and address the intended use of the device, including the needs of the user and patient. The procedures shall include a mechanism for addressing incomplete, ambiguous, or conflicting requirements. The design input requirements shall be documented and shall be reviewed and approved by a designated individual(s). The approval, including the date and signature of the individual(s) approving the requirements, shall be documented.
b) ISO 13485 Kapitel 7.3.3 („Entwicklungseingaben“)
Die ISO 13485 erwartet als Teil der Entwicklungseingaben:
- Anforderungen an die Funktion, Leistung und Sicherheit, um das Produkt der Zweckbestimmung entsprechend anwenden zu können,
- die regulatorische Anforderungen,
- ggf. Anforderungen, die sich aus vorangegangenen Entwicklungen ableiten lassen,
- Anforderungen, die sich aus dem Risikomanagement ergeben und
- sonstige Anforderungen (ohne diese näher zu spezifizieren).
Wie die FDA fordert auch die ISO 13485, dass die Produktanforderungen ermittelt, bewertet und dokumentiert werden.
c) Änderungen durch die ISO 13485:2016
Die ISO 13485:2016 hat die Anforderungen an den Design Input ergänzt.
Usability Requirements
Die Design Inputs müssen jetzt auch die „Usability Requirements“ beinhalten. Die Norm definiert nicht, was „Usability Requirements“ sind. Dazu dürften zählen:
- Nutzungsanforderungen d.h. was ein Nutzer am System eingeben, auswählen oder erkennen können muss
- Gestaltungsvorgaben wie Sie z.B. in Style Guides zu finden sind. Beispiele dafür sind Kontrastverhältnisse, Schriftgrößen und Positionen.
Verifizierbare oder validierbare Design Inputs
Bereits die Vorgängerversion der Norm verlangt, dass die Design Inputs eindeutig, widerspruchsfrei, vollständig und angemessen/korrekt sein müssen und dies durch ein Review zu überprüfen sei.
Die ISO 13485:2016 verlangt, dass die Anforderungen auch validierbar oder verifizierbar (sprich am Produkt testbar/prüfbar) sein müssen. Das sollte eigentlich selbstverständlich sein, wird jetzt aber explizit gefordert.
Entwicklungsakte
Die Design Inputs müssen Teil der Entwicklungsakte sein. Diese Akte wurde mit der ISO 13485:2016 eingeführt und entspricht dem „Design History File“ der FDA.
Lesen Sie hier mehr zum Thema Design History File.
Inhalte des Design Inputs
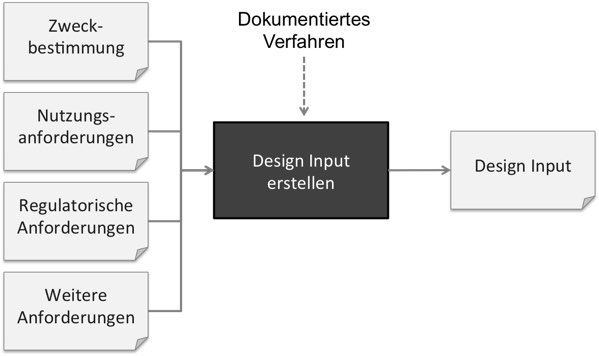
Weil Firmen häufig nicht sauber zwischen Zweckbestimmung, Erfordernissen, Stakeholder-Anforderungen und System-Anforderungen unterscheiden, wird alles als Design Input bezeichnet. Schließlich soll alles beim „Design“ beachtet werden. Doch das ist nicht korrekt.
Zweckbestimmung und Erfordernisse: Nicht Teil des Design Inputs
Die Zweckbestimmung zählt nicht zum Design Input. Die FDA fordert explizit, dass ein Verfahren dokumentiert sein muss, wie man die „Design Requirements“ (die sie synonym zu „Design Input“ verwendet) ableitet, damit diese die Zweckbestimmung erfüllen.
Die User Needs (zu deutsch Erfordernisse) zählen laut FDA noch nicht zu dem Design Input. Vielmehr muss der Design Input geeignet sein, diese Erfordernisse zu erfüllen.
Stakeholder-Anforderungen: Teil des Design Inputs
Die ISO 13485 zählt die regulatorischen Anforderungen, die zu den Stakeholder-Anforderungen zählen, explizit zu den Design Inputs.
Systemanforderungen und Systemspezifikation
Der Begriff der System Requirements Specification beinhaltet zweierlei:
- Die Systemanforderungen: Anforderungen an ein System (oft „unter der Haube“)
- Die Systemspezifikation: Festlegung, wie ein System genau gestaltet sein soll z.B. die UI-Spezifikation („über der Haube“)
Die Systemanforderungen sollte man dem Design Input zu schlagen. Schließlich bestimmen Sie die Anforderungen an das Produkt, was definitionsgemäß zum Design Input zählt.
Die System-Spezifikation (über der Haube) kann man sowohl dem Design Input zuschlagen wie dem Design Output:
- Einerseits kann man argumentieren, dass die UI-Spezifikation festlegt, wie das Produkt auszusehen hat. Dies ließe sich als Anforderungen an das Produkt und damit als Design Input deuten.
- Andererseits kann man argumentieren, dass die UI-Spezifikation bereits ein Ergebnis der Entwicklung darstellt und daher zum Design Output zählt.
Wir empfehlen die zweite Variante, weil die UI-Spezifikation in der Tat ein Entwicklungsergebnis ist.
Auch wenn die UI-Spezifikation zur Entwicklung zählt, sollte sie nicht die Aufgabe der Entwickler sein, sondern die von Usability Engineers, insbesondere Interaktionsdesignern.
Falls Sie zwischen Systemanforderungen und Systemspezifikationen nicht unterscheiden, dann schlagen Sie die System- bzw. Software-Requirements-Specification SRS dem Design Input zu.
Danke an Dr. Markus Bentrup für Hilfe bei der Präzisierung
Änderungen am Design Input
Im Lauf einer Entwicklung werden Sie den Design Input überarbeiten, entsprechend auch den Design Output. Achten Sie darauf, dass Sie alle Versionen dieser Dokumente Teil Ihres Design History Files werden lassen. Nur so können Sie die Entwicklungsgeschichte nachvollziehbar und damit glaubhaft dokumentieren.
Risikoanalyse als Design Input
Risikomanagement schlecht gemacht
Sehr oft beobachten wir folgendes Vorgehen bei Medizinprodukteherstellern:
- Medizinprodukt möglichst sicher bauen
- Risikoanalyse durchführen
- Falls inakzeptable Risiken entdeckt werden, entweder diese unter den Tisch kehren oder das Medizinprodukt sicherer machen (z.B. andere Systemarchitektur)
- Diese Maßnahmen verifizieren und alles in der Risikomanagementakte dokumentieren.
Bei diesem Vorgehen laufen Sie Gefahr, viel Zeit und Geld zu verlieren.
Besser: Risikoanalyse als Design Input
Das Ergebnis der Risikoanalyse sollte direkt Teil des Design Inputs werden.
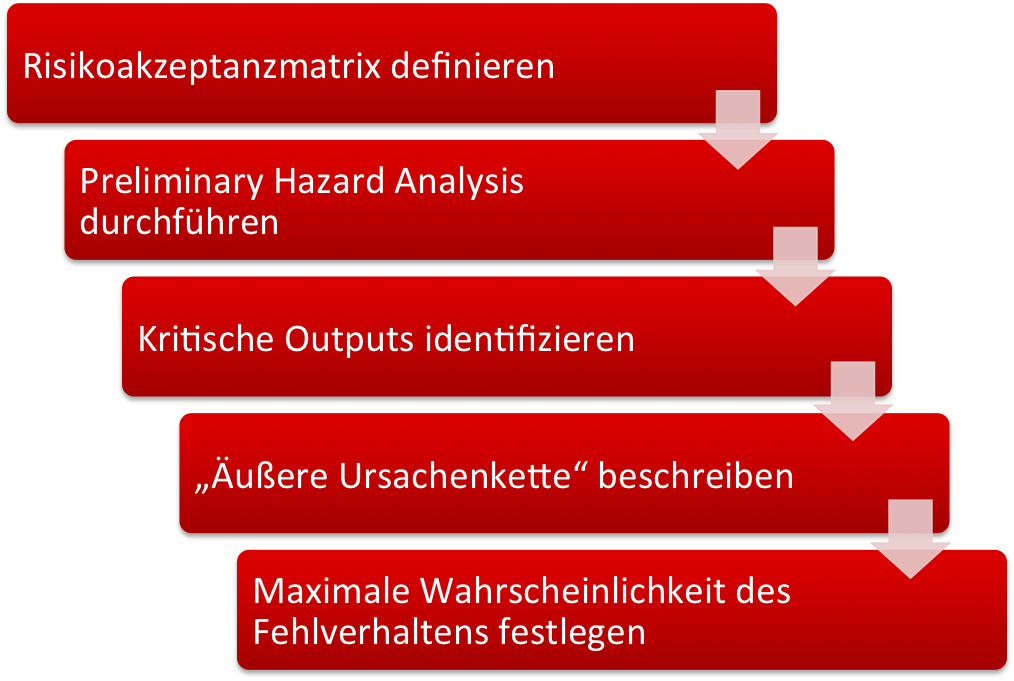
Die Risikoanalyse sollte nicht (nur) auf Basis einer Systemarchitektur erfolgen, sondern die Anforderungen in Form eines Design Inputs bestimmen.
Wir schlagen Ihnen folgendes Vorgehen vor:
- Definieren Sie quantitativ die nicht akzeptablen Risiken. (im Auditgarant lernen Sie in drei kurzen Videos, wie das geht).
- Führen Sie eine Preliminary Hazard Analysis durch und identifizieren Sie die Gefährdungen.
- Finden Sie heraus, von welchen Outputs Ihres Geräts (kann auch UI sein), welche Gefährdungen verursacht werden können.
- Beschreiben Sie, wie der Output zur Gefährdung führen kann (wenn der Output nicht bereits die Gefährdung ist) und überlegen Sie, mit welcher Wahrscheinlichkeit die Gefährdung bei einem angenommenen Fehlverhalten des Outputs des Medizinprodukts auftritt.
- Bestimmen Sie nun mit Hilfe der Risikoakzeptanzmatrix die maximale Wahrscheinlichkeit, mit der sich ein Output falsch verhalten kann. Genau diese maximale Wahrscheinlichkeit ist nun der Design Input.
Erst jetzt entwickeln Sie Ihr Medizinprodukt gemäß dieses Design Inputs. Wenn beispielsweise der Design Input eine Wahrscheinlichkeit spezifiziert, die eine Gruppe von (Elektronik)-Bauteilen oder eine selbst geschriebene Software nicht gewährleisten kann, dann werden Sie eine andere Systemarchitektur wählen, z.B. eine diversitäre.
Leider beobachten wir fast nie, dass Design Inputs (d.h. Systemanforderungen) auf konkrete Wahrscheinlichkeiten eingehen. Die Folgen sind aufwendige Nachbesserungen, verzögerte Projekte oder/und unsichere Medizinprodukte. Vermeiden Sie diese Probleme!




Ich halte den, die Risikoanalyse für quantitativen Input zu verwenden für sehr gut. Das gibt z.B. auch eine gute Brücke zu Normen der funktionalen Sicherheit mit ihren SIL-Leveln.
Ich bin bisher bei dem Vorgehen allerdings zurückhaltend, weil die MDR ja verlangt, dass das Risiko _so klein wie möglich_ ist solange das Nutzen/Risiko-Verhältnis nicht wieder ansteigt. Insofern kommt man bei jedem quantitativen Risikoakzeptanzlevel in Rechtfertigungszwang. Warum nur SIL2 anfordern, wenn doch auch SIL3 möglich wäre?
Was sind dazu die Erfahrungen? Gibt es dazu Tipps?
Sehr geehrter Herr Müller,
vielen Dank für Ihre interessante Frage!
Hier unsere Gedanken dazu: Die Risikovorgabe der MDR (alle Risiken so weit wie möglich minimieren) und die Sicherheitslevel gemäß beispielsweise IEC 61508 stellen unserer Meinung nach zweierlei Konzept dar und sollten nicht vermischt werden.
In letzterem geht es darum, die Vielzahl von Sicherheitsmaßnahmen irgendwie generisch zu clustern und über generische (!) Sicherheitslevel zur Anwendung zu bringen. Das entspricht übrigens dem „risikobasierten“ Ansatz, der sowohl von der MDR/IVDR, als auch von der ISO 13485 erwartet wird.
Das Risikomanagement nach ISO 14971 ist davon losgelöst. Hier werden ganz spezifische (!) Einzelrisiken beschrieben und wenn man diese spezifischen Einzelrisiken kennt und benennen kann, dann ist man auch verpflichtet, diese „so weit wie möglich“ zu reduzieren.
Und noch ein weiterer Gedanke, die Festlegung des SI-Levels für eine Anwendung erfolgt in der Regel mithilfe eines Risikobaums, der einer Norm für eine bestimmte Anwendung entspringt. Dies bedeutet, dass die Risiken einer Anwendung bereits bewertet wurden und das SI-Level die normative, und somit letztendlich auch die gesetzliche, Akzeptanzgrenze darstellt. Das bedeutet, die sektorspezifischen Anwendungsnormen für funktionale Sicherheit verfolgen in der Regel keinen „soweit wie möglich“-Ansatz, sondern eher einen „soweit wie nötig“-Ansatz. Dadurch hat der Hersteller einer Maschine die Möglichkeit, ein sicheres System mit SIL-zertifizierten Komponenten zu entwickeln, ohne sich mit der detaillierten Analyse von Fehler- und Schadenswahrscheinlichkeiten auseinandersetzen zu müssen, was bereits im SIL enthalten ist.
Auch für Medizinprodukte sind Sicherheitslevel in Normen festgelegt, jedoch in Form von Grenzwerten und Tests statt SI-Levels. Ein Hersteller, der nachweisen kann, dass er die Anforderungen der IEC 60601-1 erfüllt, muss nicht zwangsläufig besser sein als die Norm. Die Erfüllung der Norm ist aus rechtlicher Sicht ausreichend, da eine Norm den aktuellen Stand der Technik repräsentiert.
Ich hoffe, diese Ausführungen helfen Ihnen weiter.
Liebe Frau Bartsch,
danke für Ihre ausführliche Antwort. Auch wenn man das SIL-Konzept außen vor lässt, hat man aber ein Problem, wenn man als Output der Risikoanalyse eine konkrete Fehlerrate festlegt, die nötig ist, um zu einem akzeptablen Risiko zu kommen. Auch hier muss man sich rechtfertigen, warum man nicht einen noch tieferen Wert wählt. Auch wenn man sich auf den Stand der Technik bezieht, wie z.B. die IEC 60601-1, ist die Argumentation nicht einfach, da das dort akzeptierte Risiko nur implizit Teil der Norm ist und nirgendwo quantitativ angegben ist.
Sehr geehrter Herr Müller,
ich gebe Ihnen absolut Recht, man muss eine nachvollziehbare Argumentationslinie für die produktspezifische Risikoakzeptanz vorweisen können und das Thema ist nicht trivial. Gemäß ISO 14971 setzt sich das Risiko zusammen aus Wahrscheinlichkeit des Auftretens eines Schadens und Schweregrad des Schadens. Weiterhin fordert die Norm, die Festlegung objektiver Kriterien für die Akzeptanz von Risiken. Auch hier werden keine akzeptablen Risikobereiche festgelegt.
Das ist auch kaum generisch zu determinieren, da die Risikoakzeptanz produktspezifisch ist. Bei Festlegung der produktspezifischen Risikoakzeptanz sollten der Nutzen und mögliche Nebenwirkungen des Produkts berücksichtigt werden, sowie, wie sie auch bereits geschrieben haben, der SOTA und die unternehmensspezifische Risikopolitik. Es können hierbei mögliche Marktdaten des Produkts und basierend darauf Wahrscheinlichkeitsklassen für das Produkt sowie mögliche Schweregrade anhand beispielhafter möglicher produktspezifischer Schäden definiert werden.
Hierbei helfen vielleicht auch unsere Fachartikel zum Risikomanagement, zwei habe ich bereits im Text verlinkt, weiterhin sind folgende zum Thema interessant: Risikoanalyse und Gefährdungen und Gefährdungssituationen