
Die ISO 19011 ist der internationale Leitfaden für die Auditierung von Managementsystemen. Daher betrachtet Ihre Benannte Stelle die ISO 19011 als Stand der Technik, wenn sie bei Ihnen im ISO 13485-Zertifizierungsaudit prüft, ob Sie Ihre internen Audits und Lieferantenaudits wirksam durchführen.
Folglich sollten insbesondere die Qualitätsmanagement-Verantwortlichen die ISO 19011 kennen und berücksichtigen. Dabei hilft dieser Artikel.
Die DIN EN ISO 19011:2018-10 („Leitfaden zur Auditierung von Managementsystemen“) ist bei DIN Media und als EVS EN ISO 19011:2018 hier preisgünstiger erhältlich.
Ein Entwurf für die nächste Version der Norm (DIN EN ISO 19011 – 2025-04) ist verfügbar. Die finale Norm soll im Q1 2026 veröffentlicht werden und enthält auch das Thema KI.
1. Grundlagen der ISO 19011
1.1 Anwendungsbereich und Zweck der Norm (Kapitel 1)
Die Norm gibt Leitlinien für die Auditierung von Managementsystemen (z. B. ISO 9001, ISO 13485), ist jedoch keine harmonisierte Norm. Folglich formuliert sie keine Anforderungen, sondern dient als Handlungsempfehlung („guidance“).
Die ISO 19011 ist anwendbar in allen Organisationen, die im Rahmen eines Managementsystems interne oder externe Audits (z. B. bei Lieferanten) durchführen.
Die Zertifizierstellen selbst richten sich nach einer anderen Norm, der ISO 17021.
Die ISO 19011 verweist ausschließlich auf ISO 9000:2015 für Begriffe und Definitionen und enthält keine weiteren normativen Verweisungen. Andere Standards (wie ISO 13485) können ergänzend verwendet werden.
Die ISO 19011 hat keinen direkten Bezug auf Medizinprodukte oder IVD. Folglich beziehen sich die in der Norm genannten Chancen und Risiken nicht auf die Leistungsfähigkeit und Sicherheit der Produkte.
1.2 Aufbau der Norm
Die Norm ist in sieben Kapitel gegliedert (s. Abb. 1).
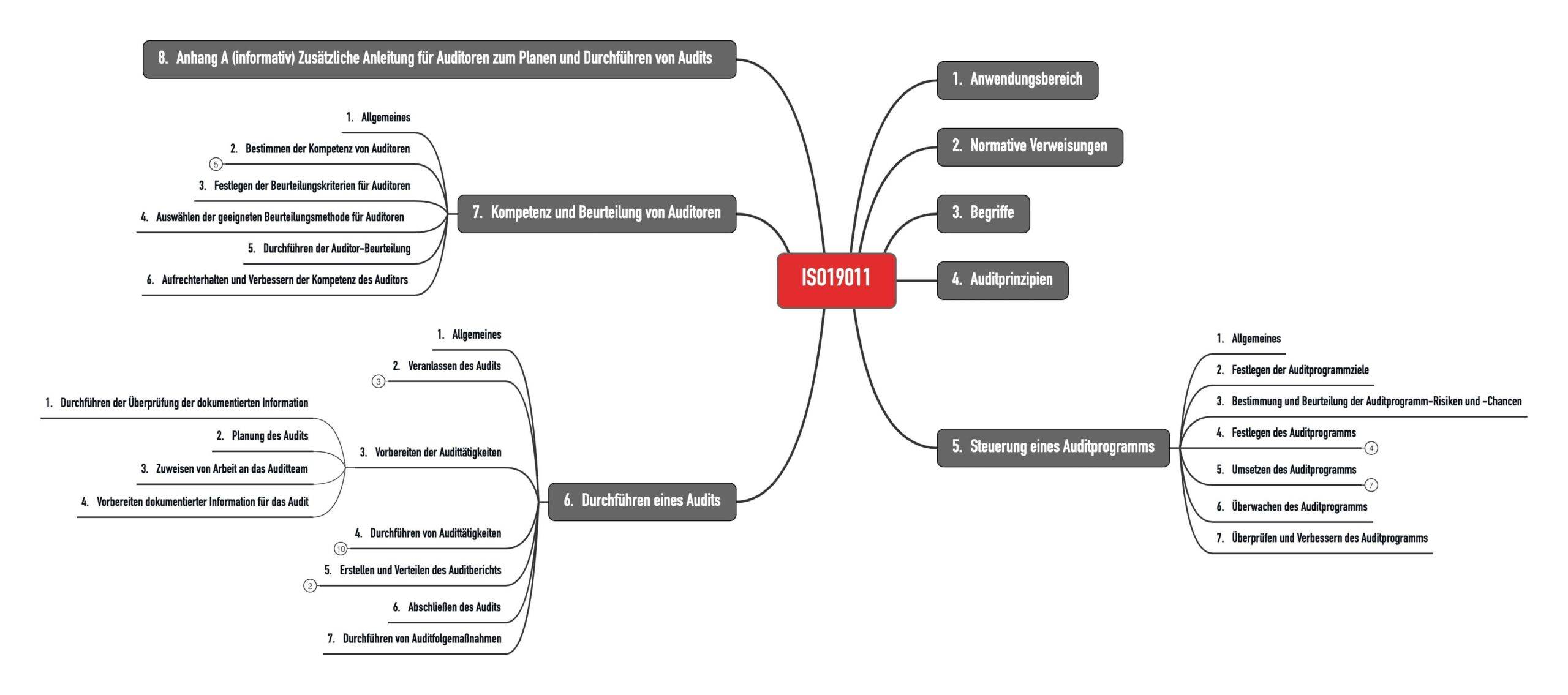
1.3 Änderungen gegenüber der Vorgängerversion
Die Ausgabe 2018 der ISO 19011 ergänzt die Vorgängerversion um den risikobasierten Ansatz und erweitert die Anleitungen sowohl für die „Verwaltung“ als auch für die Durchführung des Auditprogramms bzw. der Audits. Auch wurden die Anforderungen an die Kompetenz von Auditoren erweitert.
2. Wesentliche Inhalte der Norm
2.1 Die sieben Auditprinzipien (Kapitel 4)
Die ISO 19011 „fordert“, dass sich Organisationen beim Audit an sieben Prinzipien orientieren:
- Integrität: Grundlage für Professionalität
- Sachliche Darstellung: Verpflichtung zur wahrheitsgemäßen und genauen Berichterstattung
- Berufliche Sorgfaltspflicht: Angemessenes Urteilsvermögen bei der Auditierung
- Vertraulichkeit: Keine unbefugte Offenlegung von Informationen
- Unabhängigkeit: Basis für Unparteilichkeit und Objektivität
- Evidenzbasierter Ansatz: Methodik zur Erzielung verlässlicher Schlussfolgerungen
- Risikobasierter Ansatz: Berücksichtigung von Risiken und Chancen
2.2. Management des Auditprogramms nach ISO 19011
Die Norm legt im Kapitel 5 die Metaebene des Auditprogramms fest, d. h. dessen Steuerung – von der Zielsetzung bis zur Überprüfung des Programms.
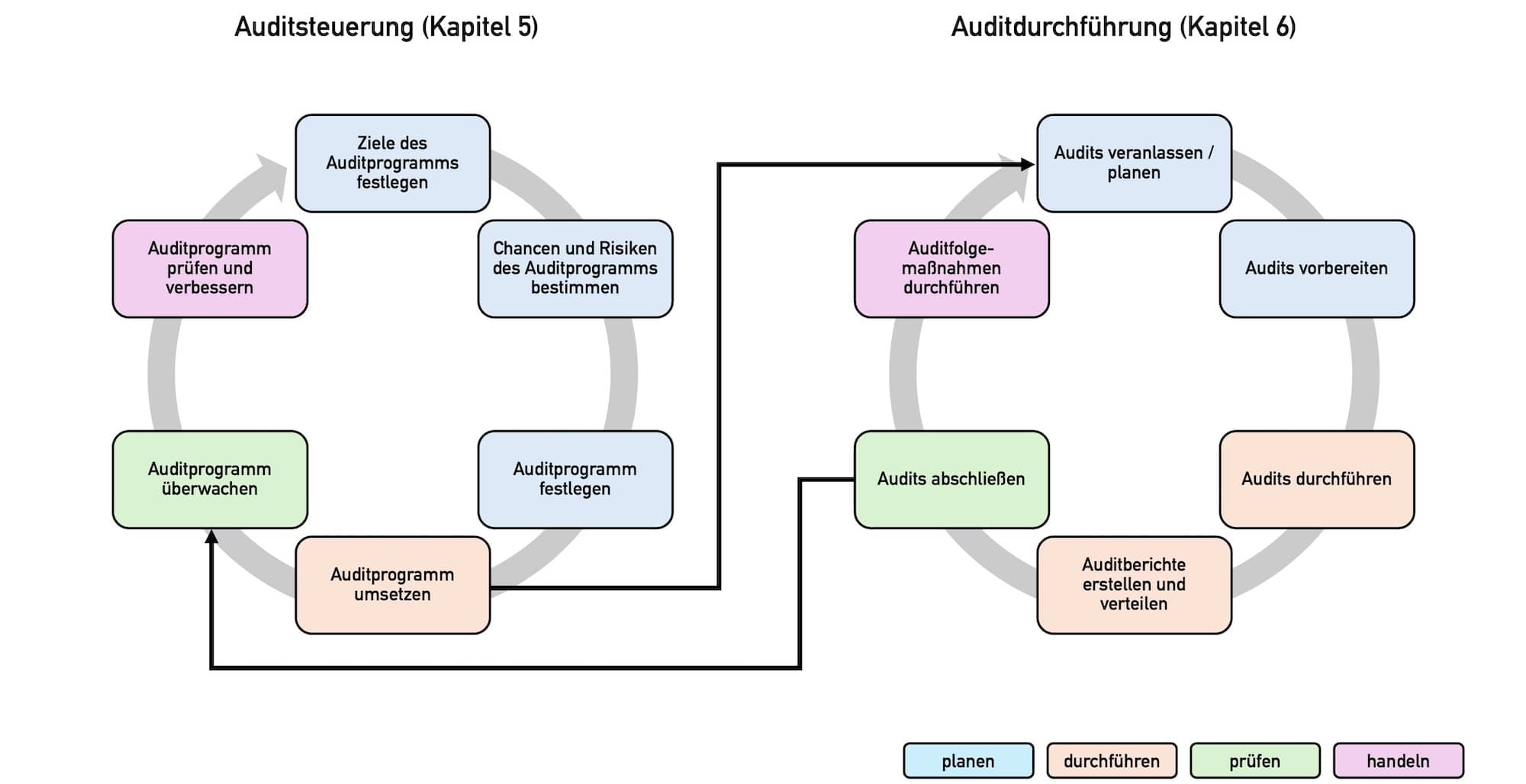
2.2.1 Zielsetzung
Die Organisationen sollen damit beginnen, die Ziele des Auditprogramms zu bestimmen (Kapitel 5.2). Diese sollten mit der Gesamtstrategie der Organisation übereinstimmen und die Leistung des Managementsystems bewerten.
Falls es Änderungen im Managementsystem gab, müssen diese bei der Zielsetzung beachtet werden. Auch müssen die Ergebnisse früherer Audits in die Zielsetzung einbezogen werden.
2.2.2 Bestimmung und Bewertung von Auditprogramm-Risiken
Im nächsten Schritt sollen die Organisationen die Chancen und Risiken des Auditprogramms identifizieren (Kapitel 5.3).
Risiken können dadurch entstehen, dass die Auditoren nicht ausreichend kompetent sind, dass die Audits keine angemessene Dauer haben, dass nicht alle Standorte auditiert werden, dass die auditierten Bereiche nicht kooperativ sind oder dass die Auditergebnisse im Anschluss nicht berücksichtigt werden.
Bei den Chancen bezieht sich die ISO 19011 v. a. auf die Effizienz. Beispiele sind, dass man mehrere Audits bei einem Besuch durchführt oder die Dauer von Reisen minimiert. Die Auswahl kompetenter Auditoren entsprechend den Auditzielen ist keine Chance, sondern eine Anforderung. Eine Chance ist hingegen, Prozesse effizienter zu gestalten, Fehler abzustellen und damit Produkte sicherer zu machen.
2.2.3 Festlegung des Auditprogramms
Mit diesen Informationen lässt sich dann das Auditprogramm planen (Kapitel 5.4). Dazu zählen:
- Rollen und Verantwortlichkeiten für Auditprogramm-Management definieren
- Umfang des Auditprogramms entsprechend Größe/Art der Organisation festlegen
- Ressourcen (finanziell, Methoden, Team etc.) bestimmen
2.2.4 Überwachung, Überprüfung und Verbesserung
Danach geht es an die Umsetzung (Kapitel 5.5) und Überwachung des Programms (5.6). Typische Tätigkeiten sollen dabei sein:
- Kennzahlen zur Überwachung des Auditprogramms definieren
- Zielerreichungsgrad des Auditprogramms regelmäßig bewerten
- Zeitplan- und Budgeteinhaltung überprüfen
- Kompetenz des Auditteams evaluieren
- Fähigkeit zur Umsetzung des Auditplans bewerten
- Feedback von Auditoren, Auditierten und anderen Parteien einholen
- Neue Praktiken/Methoden für Audits identifizieren
- Verbesserungsmöglichkeiten dokumentieren
2.3. Auditdurchführung gemäß ISO 19011
Das sechste Kapitel beschreibt konkrete, bewährte Vorgehensweisen beim Durchführen der Audits.
2.3.1 Vorbereitung der Audittätigkeiten
Das beginnt mit der Vorbereitung der Audits (Kapitel 6.2, 6.3). Überträgt man diese Anforderungen auf die Hersteller von Medizinprodukten und IVD, sollten die Organisationen auf das Folgende achten:
- Der Auditplan muss die QM-relevanten Prozesse gemäß ISO 13485 abdecken.
- Die Dokumentenprüfung soll die technische Dokumentation und relevante regulatorische Anforderungen umfassen.
- Checklisten müssen spezifische MDR-Anforderungen berücksichtigen.
- Die Stichprobenauswahl sollte auf Risikoklassen der Produkte basieren.
- Der Zugriff auf die Technische Dokumentation muss sichergestellt sein.
2.3.2 Durchführung der Audittätigkeiten
In Kapitel 6.4 geht es um die Durchführung der Audits. Für Medizinprodukte- und IVD-Hersteller würden sich bei internen Audits insbesondere folgende „Practices“ empfehlen:
- Das Eröffnungsgespräch klärt regulatorische Änderungen seit dem letzten internen Audit.
- Eine Überprüfung der Wirksamkeit des PMS/PMCF/PMPF-Systems findet statt.
- Die Verifizierung der Konformität mit grundlegenden Sicherheits- und Leistungsanforderungen wird stichprobenartig geprüft.
- Ebenso werden die Schnittstellen zu Benannten Stellen und Behörden geprüft.
- Besondere Beachtung sollten kritische Zulieferer und ausgelagerte Prozesse finden.
2.3.3 Erstellung und Verteilung des Auditberichts
Auch beim Erstellen und Verteilen des Auditberichts (Kapitel 6.5) gilt es, „Best Practices“ zu beachten, die teilweise spezifisch für den Kontext zur ISO 13485 sind.
- Feststellungen müssen regulatorische Bezüge eindeutig aufzeigen.
- Die Dokumentation muss konform mit den Anforderungen des Zertifizierers sein.
- Der Bericht wird an die relevanten Parteien verteilt.
- Den Auditbericht muss man als Teil der QM-Dokumentation archivieren.
2.3.4 Auditabschluss und Follow-up
Die Kapitel 6.6 und 6.7 beziehen sich auf das Abschließen der Audits und das Durchführen der „Auditfolgemaßnahmen“. Für Medizinproduktehersteller bedeutet dies beispielsweise:
- Korrekturmaßnahmen auf regulatorische Konformität prüfen
- Wirksamkeitsprüfung unter Berücksichtigung der MDR/IVDR-Anforderungen gewährleisten
- Berücksichtigung der Ergebnisse im Management-Review sicherstellen
- Meldepflichten (Vigilanz) beachten
- Dokumentation für mögliche Behördenaudits aufbereiten
2.4. Kompetenz und Bewertung von Auditoren
Besonderen Wert legt die ISO 19011 auf die Kompetenz des Auditorenteams (Kapitel 7).
2.4.1 Festlegung der Auditorenkompetenz
Das beginnt damit, dass die Kompetenz der Auditorinnen und Auditoren bestimmt werden muss (Kapitel 7.2.3). Dazu zählen bei Medizinprodukteherstellern:
- Nachgewiesene Kenntnisse der ISO 13485 und MDR/IVDR
- Verständnis der grundlegenden Sicherheits- und Leistungsanforderungen
- Kenntnis der harmonisierten Normen und Common Specifications
- Verständnis der Besonderheiten der verschiedenen Produktkategorien
- Erfahrung mit Klassifizierungsregeln für Medizinprodukte
- Verständnis des Risikomanagements nach ISO 14971
- Kenntnisse der klinischen Bewertung/Leistungsbewertung und PMS/PMCF/PMPF
2.4.2 Kompetenzkriterien
Diese Kompetenzen müssen die Organisationen auch nachweisen (Kapitel 7.2.3, 7.2.4). Das kann im Medizinprodukteumfeld beispielsweise gelingen durch:
- Berufserfahrung im Medizinproduktebereich
- Nachweise über spezifische Medizinprodukteausbildung
- Erfahrung in vollständigen Audits im MP-Bereich
- Tätigkeiten bei z. B. einer Benannten Stelle oder Überwachungsbehörde
- Teilnahme an Seminaren mit Wirksamkeitsnachweis
Nicht nur, aber auch weil es die Norm verlangt, sollten Hersteller ihre internen Auditoren systematisch ausbilden. Ziehen Sie das Seminar „Interner Auditor“ vom Johner Institut in Betracht.
2.4.3 Bewertungsmethoden
Neben den formalen Voraussetzungen sollten die Auditoren selbst fortlaufend beobachtet und bewertet werden (Kapitel 7.3, 7.4):
- Beobachtung während Audits mit regulatorischem Fokus
- Überprüfung der Auditberichte auf regulatorische Vollständigkeit
- Bewertung der Fähigkeit, regulatorische Abweichungen zu erkennen
- Evaluation der Kommunikation mit der Rolle QMB und dem Regulatory Affairs Team
- Beurteilung des Verständnisses von Zulassungsprozessen
- Prüfung der Kompetenz in verschiedenen Produktkategorien
2.4.4 Aufrechterhaltung und Verbesserung der Kompetenz
Auch unabhängig von diesen Ergebnissen empfiehlt es sich, die Kompetenz kontinuierlich auszubauen oder zu aktualisieren (Kapitel 7.6):
- Regelmäßige Teilnahme an Schulungen zu regulatorischen Änderungen
- Teilnahme an Workshops zu MDR-/IVDR-spezifischen Themen
- Kontinuierliche Aktualisierung des Wissens zu harmonisierten Normen
- Erfahrungsaustausch mit anderen Auditoren im MP-Bereich
All diese Weiterbildungen und deren Wirksamkeit müssen die Organisationen dokumentieren.
3. Fünf Tipps zur Umsetzung
Tipp 1: Interne Audits der internen Audits nicht vergessen
Die ISO 19011 hilft dabei, interne Audits zu planen und durchzuführen. Aber auch die Wirksamkeit der internen Audits selbst sollte überprüft werden. Das geschieht durch interne Audits der internen Audits ebenso wie im Rahmen des Management-Reviews. Entsprechend sollte die ISO 19011 bei beidem berücksichtigt werden.
Tipp 2: Auditprogramm risikobasiert gestalten
Der ISO 19011 fehlt der Bezug zu Medizinprodukten bzw. IVD und damit zur Patientensicherheit. Die Hersteller sollten dem risikobasierten Ansatz der ISO 19011 folgen, dabei aber die Risiken im Sinne der ISO 14971 im Blick behalten.
Die ISO 19011 empfiehlt, die Risiken durch fehlende Kompetenz von Auditorinnen und Auditoren zu betrachten. Medizinproduktehersteller sollten die Konsequenzen für die Patienten betrachten, die sich aus den Kompetenzmängeln ergeben könnten.
Tipp 3: Nicht nur die Konformität prüfen
Mit den Audits sollten die Organisationen mehr erreichen, als nur ihren gesetzlichen Pflichten gerecht zu werden. Sie können die Audits auch nutzen, um
- die Wirksamkeit der Prozesse zu bestimmen,
- Ineffizienzen in den Prozessen zu entdecken und zu beseitigen,
- Verfahrens- und Arbeitsanweisungen zu entschlacken,
- Schwachpunkte bei den Zulieferern zu entdecken (und ggf. bessere zu wählen) und
- das Wissen in der Organisation über die Organisation und deren Produkte zu fördern.
Tipp 4: Ergebnisse vielfältig nutzen
Zudem können den Unternehmen die Ergebnisse der Audits dienlich sein
- um Audits durch die eigenen Kunden zu minimieren (besonders bei Zulieferern relevant),
- als Input für Management-Reviews und das CAPA-System,
- zur Verbesserung der Produkte,
- als Informationen für das Risikomanagement und die Post-Market Surveillance,
- als Nachweis der regulatorischen Anforderungen.
Tipp 5: Spezifische Anforderungen im Blick behalten
Weil die ISO 19011 „domänenagnostisch“ ist, sollten die Medizinprodukte- und IVD-Hersteller die spezifischen Anforderungen im Blick behalten. Diese betreffen beispielsweise:
- Aufbewahrungspflichten von Unterlagen.
- Meldepflichten an Behörden und Benannte Stellen bei Auditfeststellungen.
- Spezifische Kompetenzanforderungen.
- Anforderungen der ISO 13485 und MDR/IVDR an das QM-System.
4. Fazit und Zusammenfassung
Die Norm ist ein sehr nützlicher Leitfaden, der hilft, die Anforderungen der ISO 13485 an die Planung des Qualitätsmanagementsystems und an interne Audits sowie Lieferantenaudits zu erfüllen.
Auch deshalb ist die ISO 19011 eine Pflichtlektüre für alle Personen, die Audits planen, vorbereiten und durchführen. Das sind nicht nur die Qualitätsmanagementbeauftragten, sondern beispielsweise auch die bei den Audits beteiligten Prozessverantwortlichen.
Die ISO 19011 wurde im Vergleich zu ihrer Vorgängernorm um den risikobasierten Ansatz erweitert. Medizinproduktehersteller sollten bei diesen Risiken explizit regulatorische Risiken und Risiken für die Sicherheit der Produkte betrachten und nicht nur die von der Norm genannten Risiken.
Die Wirksamkeit von Qualitätsmanagementsystemen steht und fällt mit der Kompetenz der Auditorinnen und Auditoren. Deshalb widmet die Norm diesem Aspekt ein ganzes Kapitel.
Das Johner Institut hilft Herstellern von Medizinprodukten und IVD sowie Betreibern beim Aufbau, Prüfen und Optimieren von QM-Systemen.
Seminare wie Interner Auditor, Zertifizierter ISO 13485 Lead Auditor, Beschaffung und Lieferantenmanagement sowie Grundlagenseminar ISO 15189 vermitteln die geforderten Kompetenzen.
Änderungshistorie
- 2025-05-15: Hinweis auf neue Version der Norm oben im Artikel eingefügt
- 2025-05-13: Artikel vollständig neu geschrieben und dabei die 2018er-Version der ISO 19011 berücksichtigt
- 2016-01-12: Erste Version des Artikels veröffentlicht


Guten Tag Herr Prof. Johner,
unsere benannte Stelle möchte von Jahr zu Jahr mehr Dokumente, schon vorab eines Audits, als digitale Version zur Einsicht erhalten. Früher wurde nur das Qualitätsmanagement-Handbuch gefordert. Mittlerweile sind Qualitätssicherungsrichtlinien, Standardverfahrensanweisungen und sogar Arbeitsprotokolle und Produktionsdokumentationen dazu gekommen.
Gibt es eine Liste welche Dokumente vorab von der benannten Stelle angefordert werden können?
Beste Grüße,
Dr. Andreas Breß
Sehr geehrter Herr Dr. Breß,
vielen Dank für Ihre wertvolle Anfrage.
Ich gehe davon aus, dass Ihre Benannte Stelle nicht nur das QMS nach ISO 13485 sondern auch Ihr(e) Produkt(e) nach der EU-Verordnung 2017/45 (MDR) prüft. Mittlerweile gehen die Notified Bodies dazu über, die Dokumente der technischen Dokumentation (MDR, Anhang II) vorab anzufordern. Sie sollten also die technische Dokumentation, soweit vorab gefordert, bei Ihrer Benannten Stelle einreichen.
Sehr geehrter Herr Prof. Johner,
vielen dank für die Informationen. Wir stellen IVDs her, fallen also erst in Zukunft vollständig unter die IVDR. Bis jetzt dürfen wir noch alle unsere Produkte ohne Beteiligung einer Benannten Stelle vertreiben. PMS ist natürlich jetzt schon gefragt aber diese Dokumente wollte die Benannte Stelle gar nicht im Vorweg einsehen. Natürlich wird sich dies mit der IVDR sich ändern. Ich war nur überrascht, dass die Benannte Stelle jetzt schon mit diesem Prozedere anfängt.
Sehr geehrtes Johner-Team,
in unserer Firma gibt es Unklarheiten darüber, wie die Auditorenkompetenz definiert werden soll. Einige sind der Meinung, dass die Schulung zur ISO 19011 ausreicht, da das Zertifikat auch auf die 13485 verweist. Der Schwerpunkt lag also in dieser Richtung. Die andere Meinung lautet, dass die Kompetenz in einer separaten Schulung zur ISO 13485 erworben werden sollte. Was ist nun richtig? Wie argumentiert eine Benannte Stelle hier? Nicht, dass dies zu einer Abweichung im Audit führt.
Vielen Dank.
Sehr geehrter Herr Sammer,
das ist eine ausgezeichnete Frage! Vielen Dank!
Es gibt keine konkrete Forderung, nach einer der beiden Normen geschult zu sein. Aber es gibt die Forderung, die Kompetenzen für die Tätigkeiten nachweisen zu müssen, um die Aufgaben innerhalb des QMS zu erledigen.
Wenn beispielsweise der Auditor oder die Auditoren die Auditplanung macht, dann ist eine Schulung gemäß den Vorgaben der ISO 19011 empfehlenswert. Wenn die gleiche Person den Umgang mit CAPAs auditiert, sollte sie die Anforderungen der ISO 13485 daran verstanden haben und entsprechend geschult sein.
In anderen Worten: Die Antwort hängt von den Aufgaben der Auditoren ab.
Beste Grüße, Christian Johner