Weil die Güte der Sterilisation von Medizinprodukten entscheidend für die Sicherheit dieser Medizinprodukte ist, sind die Auswahl und die Validierung des Sterilisationsverfahrens regelmäßig Gegenstand von Audits und Inspektionen.
Dieser Artikel verschafft einen schnellen Überblick über die verschiedenen Sterilisationsverfahren sowie die regulatorischen Anforderungen bei Medizinprodukten und liefert Best Practices, um Prüfungen durch Behörden und Benannte Stellen sicher zu bestehen.
1. Sterilisation: Der Kontext
a. Definition
Unter Sterilisation versteht man in Medizin und Hygiene ein validiertes Verfahren, um Medizinprodukte, Instrumente oder Materialien in einen Zustand völliger Keimfreiheit zu versetzen. Dabei werden alle Formen mikrobiellen Lebens, einschließlich Bakterien, Viren, Pilze und deren Sporen, abgetötet oder entfernt.
Medizinprodukte gelten als keimfrei, wenn die Wahrscheinlichkeit, dass ein lebensfähiger Mikroorganismus auf dem Produkt vorhanden ist, unter 10−6 liegt. Dies bedeutet normativ (DIN EN 556), dass weniger als ein Mikroorganismus pro einer Million Einheiten (SAL-Wert: Sterility Assurance Level 10−6) vorhanden sein darf. Dann gilt das Medizinprodukt als steril.
Die Sterilisation wiederverwendbarer Medizinprodukte ist ein Sonderfall der Aufbereitung von Medizinprodukten.
b. Beispiele
- Medizinprodukte, die in Kontakt mit nicht intakter Haut gelangen (z. B. sterile Wundkompressen) oder gar implantiert werden, werden meist vom Hersteller steril in Verkehr gebracht.
- Bei wiederverwendbaren Produkten wie chirurgischen Instrumenten liegt die Verantwortung für die Sterilisation hingegen beim Betreiber (Krankenhäuser, Praxen). Sowohl Hersteller als auch Betreiber können hierfür auf externe Sterilisationsdienstleister zurückgreifen.
- Keine Sterilisation erfordern dagegen Produkte, die ausschließlich mit intakter Haut in Kontakt kommen (z. B. Gehhilfen) oder keinen Patientenkontakt haben (z. B. Software).
Teilweise greifen sowohl Hersteller als auch Betreiber auf Sterilisationsdienstleister zurück.
2. Sterilisationsverfahren
a. Übersicht
Um Medizinprodukte zu sterilisieren, stehen mehrere Verfahren zur Verfügung, die sich je nach Anwendungsfall besser oder schlechter eignen.
| Verfahren | Prinzip | Besonderheiten | Beispiele |
| Dampfsterilisation (Feuchte Hitze) | Gesättigter Wasserdampf bei hoher Temperatur und Druck | Sehr robustes, etabliertes Verfahren Nicht geeignet für hitze- oder feuchtigkeitsempfindliche Materialien | Chirurgische Instrumente aus Metall, Textilien (OP-Tücher/Kleidung), wiederverwendbare MPs |
| Ethylenoxid-Sterilisation (EO) | Gassterilisation bei niedrigen Temperaturen | Sehr gute Durchdringung poröser Materialien Rückstände (EO, ECH, Ethylenglykol) müssen kontrolliert werden Lange Prozess- und Belüftungszeiten | Katheter und Schläuche, Produkte aus Kunststoffen und Elastomeren, Textilien mit Elastan/Polymeren, komplexe Baugruppen mit Hohlräumen, Medizinprodukte mit Elektronik |
| Gamma-Sterilisation | Ionisierende Strahlung (Cobalt-60) | Industrielles Verfahren, hohe Durchsatzraten Kann Materialalterung bzw. Veränderung sowie Verfärbung verursachen Materialabhängig | Einmalspritzen, Kanülen, OP-Handschuhe, Implantate, verpackte Einmalprodukte, Nahtmaterial |
| Elektronenstrahl-Sterilisation (E-Beam) | Beschleunigte Elektronen | Kürzere Prozesszeiten als Gamma Geringere Eindringtiefe → Einschränkungen bei dicken/komplexen Produkten | Einmalartikel mit geringer Materialdicke, Spritzen, Tubing, flache Kunststoffprodukte, verpackte Einwegprodukte |
| Niedertemperatur-Plasma (z. B. Wasserstoffperoxid-Plasma) | Reaktive Plasma-Spezies bei niedriger Temperatur | Kurze Zyklen Eingeschränkte Material- und Geometriekompatibilität Vor allem für Wiederaufbereitung, weniger für industrielle Serienfertigung | Endoskope (eingeschränkt, je nach System), temperaturempfindliche Instrumente, optische Komponenten |
Wenn von chemischer Sterilisation gesprochen wird, dann ist in der Regel eine Sterilisation mit Ethylenoxid (EO) gemeint. Andere chemische Verfahren bilden im Zusammenhang mit Medizinprodukten Sonderfälle (z. B. Peroxid).
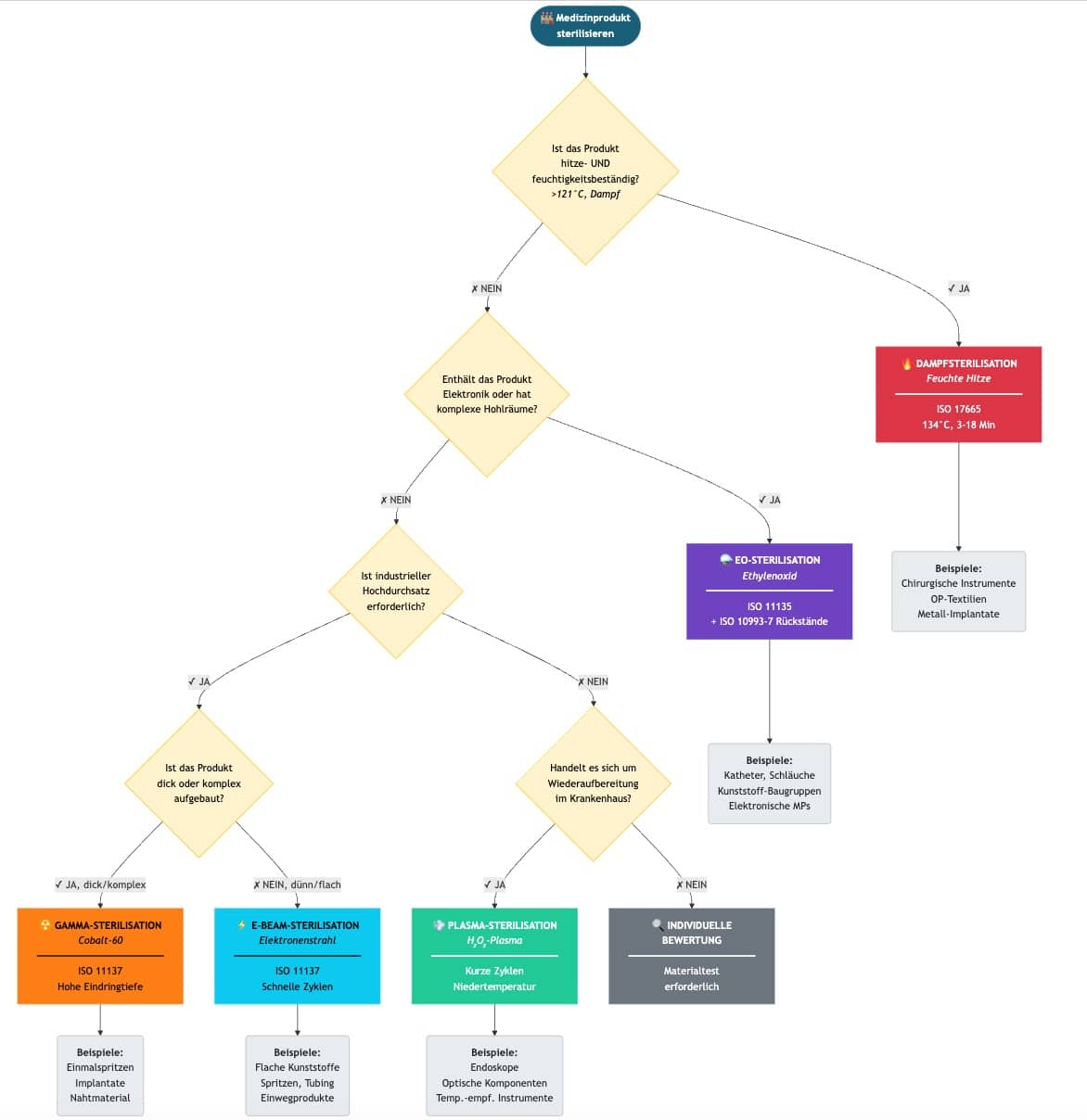
b. Auswahl
Die Auswahl eines Sterilisationsverfahrens hängt von den oben genannten Attributen ab. Hersteller können ihre Auswahl in Form eines Entscheidungsdiagramms dokumentieren, wie es beispielhaft die Abb. 1 skizziert.
3. Regulatorische Anforderungen an die Sterilisierung
a. Anforderungen der MDR
Die Anforderungen der MDR zum Thema Sterilisation finden sich im Anhang I, Kapitel II, Abschnitt 11.1:
„Produkte, die in sterilem Zustand in Verkehr gebracht werden, müssen unter geeigneten und validierten Verfahren hergestellt und sterilisiert werden.“
b. Normative Anforderungen
Den Nachweis, dass diese regulatorischen Anforderungen eingehalten werden, erbringen Hersteller auch dadurch, dass sie relevante (harmonisierte) Normen beachten.
| Norm | Inhalt |
| ISO 11135 | EO-Sterilisation |
| ISO 17665 | Feuchte Hitze/Dampfsterilisation |
| ISO 11137-1/-2/-3 | Strahlensterilisation |
| ISO 11138-Serie | Biologische Indikatoren |
| ISO 11140-Serie | Chemische Indikatoren |
| EN 556-1 | Anforderungen für „STERIL“-Kennzeichnung |
| ISO 10993-7 | EO-Rückstände |
Anforderungen zur Prozessvalidierung nennt auch die ISO 13485.
Beachten Sie auch unseren Fachartikel ISO 17664 und Aufbereitung von Medizinprodukten.
c. Anforderungen der FDA
Anforderungen an die Hersteller
Bei 510(k)-Einreichungen für sterile Produkte müssen detaillierte Beschreibungen des Sterilisationsprozesses, der Verpackung und der Validierung vorgelegt werden. Änderungen am Sterilisationsverfahren erfordern oft eine neue Bewertung oder eine neue 510(k)-Einreichung.
Die FDA betont zudem die Notwendigkeit, bei der Auswahl des Sterilisationsverfahrens Umweltauswirkungen zu berücksichtigen, während die Sicherheit für die Patienten oberste Priorität hat.
Die FDA informiert über die wichtigsten Anforderungen an die Sterilisation von Medizinprodukten. Sie verweist dort auch auf die oben genannten Normen.
Anforderungen an die Lohnsterilisierer
Unternehmen, die Lohnsterilisationsleistungen (Contract Sterilization) anbieten, sind verpflichtet, ihre Einrichtungen jährlich bei der FDA zu registrieren.
4. Tipps zur gesetzeskonformen Sterilisation von Medizinprodukten
a. Strukturiert vorgehen
Wie bei jedem Prozess, gilt es auch bei der Sterilisation von Medizinprodukten, strukturiert vorzugehen:
- Zweckbestimmung und bestimmungsgemäßen Gebrauch des Medizinprodukts präzise definieren. Dazu zählen:
- Lebensdauer des Produkts
- Qualifizierung der Anwender (auch der für die Sterilisation verantwortlichen Personen)
- Anzahl der Wiederaufbereitungen
- Anforderungen an das sterilisierte Medizinprodukt
- Geeignetes Sterilisationsverfahren auswählen und Parameter bestimmen
- Berücksichtigung der Materialstabilität mit dem gewählten Verfahren
- Geeignetes Verpackungssystem festlegen
- Sterilisationsverfahren validieren
- Validierungsplan erstellen
- Validierungs-Infrastruktur etablieren
- Validierung durchführen
- Validierungsbericht erstellen
b. Kompetenzen sicherstellen
Hersteller sollten die Auswahl und Validierung des Sterilisationsverfahrens mit einem interdisziplinären Ansatz verfolgen. Je nach Verfügbarkeit empfiehlt es sich, Fachleute einzubeziehen, u. a. aus den Bereichen Materialwissenschaft, Biokompatibilität, Sterilisation und Produktentwicklung, um technische, biologische und regulatorische Anforderungen ganzheitlich abzudecken.
c. Parametrische Freigabe in Betracht ziehen
Die parametrische Freigabe ermöglicht es, auf kosten- und zeitintensive batchabhängige Sterilitätstests zu verzichten. Voraussetzung hierfür sind validierte Prozessparameter, die eine zuverlässige Steuerung und Reproduzierbarkeit des Sterilisationsprozesses sicherstellen. Robuste Daten zur mikrobiologischen Grundbelastung können eine entscheidende Rolle spielen.
d. Spezialbedingungen der jeweiligen Sterilisationsmethode beachten
Jede Sterilisationsmethode stellt spezifische Anforderungen an Material, Verpackung und Prozessführung. So erfordert etwa die EO-Sterilisation eine gezielte Steuerung von Feuchte, Temperatur und Gaskonzentration sowie eine anschließende Desorptionsphase zur Minimierung toxischer Rückstände. Bei der Strahlensterilisation hingegen stehen Materialverträglichkeit und Dosisverteilung im Vordergrund, während die Dampfsterilisation insbesondere die Thermostabilität des Produkts und der Verpackung voraussetzt.
5. FAQ
a. Welches Sterilisationsverfahren ist für Kunststoffe geeignet?
Für Kunststoffe eignen sich primär die EO- und die Strahlensterilisation, da beide Verfahren bei niedrigen Temperaturen arbeiten und thermisch empfindliche Materialien schonen. Die Dampfsterilisation ist nur bei thermostabilen Kunststoffen einsetzbar.
Entscheidend für die Verfahrenswahl sind Materialverträglichkeit, Produktgeometrie und regulatorische Anforderungen, die im Rahmen der Produktentwicklung und der Prozessvalidierung berücksichtigt werden.
b. Wie oft muss eine Sterilisationsvalidierung wiederholt werden?
Eine vollständige Revalidierung ist regulatorisch nicht in festen Zeitintervallen vorgeschrieben, wird jedoch bei wesentlichen Änderungen an Produkt, Verpackung oder Prozess gefordert.
Die einschlägigen Normen (ISO 11135, ISO 11137, ISO 17665) verlangen zudem mindestens eine jährliche Routineüberwachung zur kontinuierlichen Bestätigung des validierten Zustands (Bestandsprodukten).
Direkt nach einer Sterilisationsvalidierung (Neuprodukt/Neuer Prozess) ist zunächst eine quartalsweise Überprüfung über mindestens ein Jahr erforderlich, um saisonale Einflüsse zu berücksichtigen. Bei nachgewiesener stabiler und robuster Prozessleistung kann die Frequenz anschließend auf halbjährliche und später auf eine mindestens jährliche Überwachung reduziert werden.
Ergänzend fordern ISO 13485 und die MDR eine regelmäßige Bewertung der Prozessfähigkeit im Rahmen des Qualitätsmanagementsystems.
c. Was sind typische Fehler, die in Audits auffallen?
- Lückenhafte oder veraltete Validierungsdokumentation: Protokolle, Berichte oder Masterpläne entsprechen nicht dem aktuellen Prozessstand.
- Unzureichende Routineüberwachung: Fehlende oder verspätete Dosisaudits, Bioburden-Reviews oder Prozessparameterkontrollen
- Mangelhaftes Change-Control-Management: Änderungen an Produkt, Verpackung oder Beladung werden nicht systematisch auf den validierten Zustand bewertet.
- Fehlende oder unvollständige Risikobewertung: Keine nachvollziehbare Verknüpfung zwischen Risikomanagement (ISO 14971) und Validierungsaktivitäten
- Fehlerhafte oder unzureichende Begründungen für Worst-Case-Auswahl oder Bildung von Produktfamilien
- Defizite bei der Lieferantenqualifizierung: Unzureichende vertragliche Vereinbarungen und fehlende regelmäßige Audits externer Sterilisationsdienstleister
d. Worauf sollte man bei der Auswahl von Lohnsterilisierern achten?
Bei der Auswahl von Lohnsterilisierern helfen folgende Kriterien:
- Zertifizierung, Vollständigkeit des QM-Systems
- Einbindbarkeit in das eigene QM-System
- Erfahrung mit dem geforderten Sterilisationsverfahren
- Erfahrungen mit den Produktklassen und deren Besonderheiten (z. B. Geometrien, Materialien)
- Räumliche Nähe, logistische Anbindung
- Durchlaufzeiten
- Reporting, Transparenz (z. B. Statistiken), Prozessaufzeichnungen
- Größe, Historie, finanzielle Stabilität
- Preis
5. Fazit und Zusammenfassung
Die Verfahren zur Sterilisation von Medizinprodukten sind meist bewährt. Dennoch stoßen Behörden und Benannte Stellen oft auf Schwierigkeiten. Diese werden überwiegend dadurch verursacht, dass die Medizinproduktehersteller die regulatorischen Anforderungen nicht beachten, die Spezifika ihrer Produkte nicht berücksichtigen oder gegen Best Practices verstoßen.
Dieser Artikel hat gezeigt, wie Hersteller durch strukturiertes Vorgehen und die Sicherstellung von Kompetenzen diese Schwierigkeiten vermeiden können.
Die Expertinnen und Experten des Johner Instituts unterstützen Medizinproduktehersteller beispielsweise durch ein Review bestehender Validierungen. So stellen sie z. B. sicher, dass bei der EO-Sterilisation die Rückstände gem. ISO 10993-7 überprüft und toxikologisch bewertet sind.
Dazu betrachten die Experten den Validierungsprozess auch aus der Biokompatibilitäts-Perspektive und sorgen dafür, dass die regulatorisch geforderten Nachweise vollständig und konform vorliegen und die Patientensicherheit gewährleistet wird.