
Die 510(k)-Zulassung ist das wichtigste Verfahren, um Medizinprodukte in den USA zuzulassen. Es wird auch als Premarket Notification (PMN) bezeichnet.
Dieser Artikel verschafft eine schnelle Übersicht über das Verfahren und die Dokumente, die einzureichen sind.
Beachten Sie eine Übersicht über die Fachartikel zur FDA und deren Zulassungsverfahren.
1. 510(k)-Zulassung: Grundlagen
Im Gegensatz zum europäischen Rechtsraum kennt die FDA eine explizite Zulassung von Medizinprodukten. Das prominenteste Zulassungsverfahren ist nach einem Artikel des Food, Drug and Cosmetic Acts (FD&C) benannt: 510(k).
Dieses Zulassungsverfahren können Hersteller in der Regel dann einsetzen, wenn ihre Produkte in die Klasse II fallen und es ein vergleichbares, bereits zugelassenes Produkt gibt. Dieses Produkt, das sogenannte Predicate Device, muss auch tatsächlich vergleichbar sein. Man spricht von substantially equivalent.
Erfahren Sie weiter unten mehr über „Predicate Devices“.
Das 510(k)-Verfahren ist auch anwendbar für gewisse Klasse I-Produkte, die nicht „510(k)-exempt“ sind, sowie für eine geringe Anzahl an Klasse III-Produkten.
Die 510(k)-Varianten
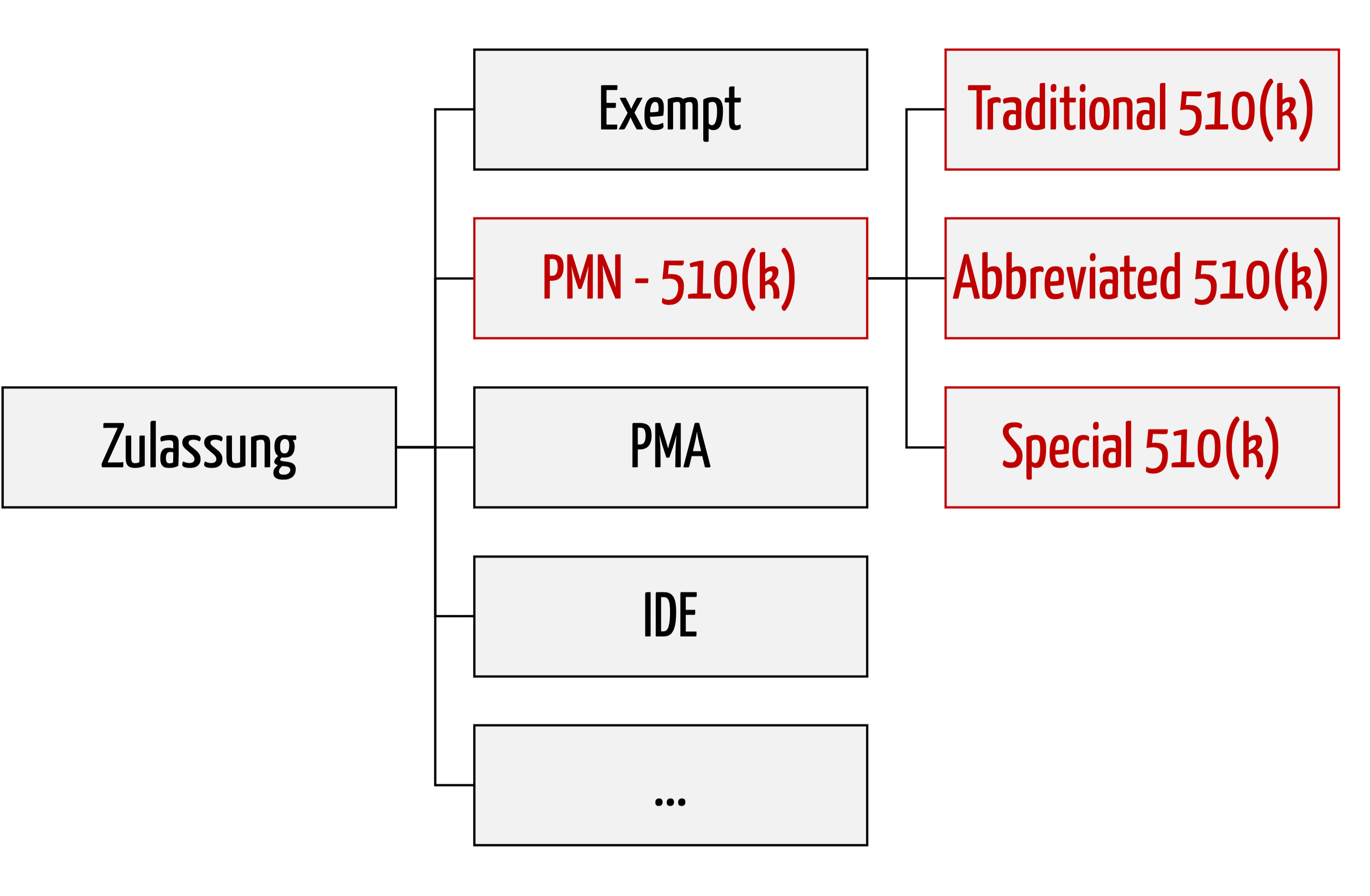
Bei den Premarket Notifications unterscheidet die FDA drei Varianten:
- Traditional 510(k)
Das klassische Zulassungsverfahren, das bei „me too“-Produkten zum Einsatz kommt und als Fallback dient, wenn die FDA Special 510(k) und Abbreviated 510(k) nicht anerkennt. - Special 510(k)
Special 510(k) ist ein beschleunigtes Verfahren, das bei Änderungen am eigenen Produkt zur Anwendung kommen kann. - Abbreviated 510(k)
Abbreviated 510(k) ist ein Verfahren, das auf die Einhaltung und Befolgung von Normen, „Special Controls“ und „Guidance Documents“ (Leitlinien) baut. Eine Sonderform dieses Verfahrens ist der neuartige Safety and Performance Based Pathway.
Lesen Sie hier mehr zum Thema Special 510(k) und Abbreviated 510(k).
2. Inhalt und Format der Submission-Dokumente
Um die Effizienz des 510(k)-Verfahrens zu erhöhen, hat die FDA inzwischen ein elektronisches Einreichungsformat eingeführt, das sogenannte eSTAR-Format. Die FDA akzeptiert zum 01.10.2023 510(k)-Einreichungen nur noch per eSTAR. Es handelt sich dabei um eine interaktive Vorlage im PDF-Format. Diese deckt die Anforderungen des 21 CFR part 807 Subpart E ab und orientiert sich dabei an den Vorgaben des IMDRF, genauer gesagt dem „Table of Contents“-Format. Zu jedem Abschnitt in der Vorlage ist die Nummer des entsprechenden Kapitels der Table of Contents-Struktur angegeben. Dies ist sehr zu begrüßen und bringt uns, zumindest einen kleinen Schritt, Richtung Harmonisierung eines weltweiten Einreichungsformats näher. Schade, dass die EU-Kommission sich bei Anhang II der MDR zur technischen Dokumentation nicht ebenfalls daran orientiert hat.
Das eSTAR-Template zum 510(k) (non-IVD) folgt dieser Struktur:
- General Introduction (u.a. Versionshistorie des Templates)
- Application/Submission Type (z.B. traditional, abbreviated, special)
- Administrative Information (Kontaktdaten, Referenz zu Pre-Submissions, Liste der angewandten Normen)
- Device Description (ausführliche Produktbeschreibung)
- Indications for Use (Statement zur Zweckbestimmung bzw. den Indikationen des Produkts)
- Classification (Informationen zur Klassifizierung des Produkts einschließlich Regulation Number und Product Code)
- Marketing History (Informationen zur Markthistorie)
- Predicates and Substantial Equivalence (ausführlicher Vergleich mit dem oder den ausgewählte(n) Predicate Devices)
- Labeling (Kennzeichnung und Gebrauchsanweisung)
- Reprocessing, Sterility, and Shelf-Life (Informationen zur Aufbereitung, Sterilisation und Shelf-Life, soweit anwendbar)
- Biocompatibility (Informationen zur Bewertung der Biokompatibilität, soweit anwendbar)
- Software/Firmware and Cybersecurity/Interoperability (SW-Dokumentation einschließlich Cybersecurity und Informationen zur Interoperabiliät, soweit anwendbar)
- EMC, Wireless, Electrical, Mechanical and Thermal Safety (Nachweise, wie z.B. Prüfberichte zu den genannten Aspekten, soweit anwendbar)
- Performance Testing (Nachweise zu weiteren Tests, wie „bench“-, Tier- oder klinischen Tests)
- References (z.B. zu referenziertes Fachliteratur)
- Administrative Documentation (u.a. Truthful and Accurate Statement, Declaration of Conformity, 510(k) Summary oder Statement)
Hinweise darauf, was die einzelnen Kapitel enthalten sollen, finden Sie in der Leitlinie Electronic Submission Template for Medical Device 510(k) Submissions.
3. Predicate Device
Beachten Sie den ausführlicheren Artikel zu „Predicate Devices“ und zur „Substantial Equivalence“.
Ein Predicate Device ist ein bereits (legal) in den USA vermarktetes Medizinprodukt, auf das Medizinproduktehersteller bei einer Zulassung verweisen können. Solch ein Predicate Device ist eine Voraussetzung für das von den Herstellern bevorzugte 510(k)-Zulassungsverfahren, das im Vergleich zu einer PMA (Premarket Approval) weniger aufwendig ist.
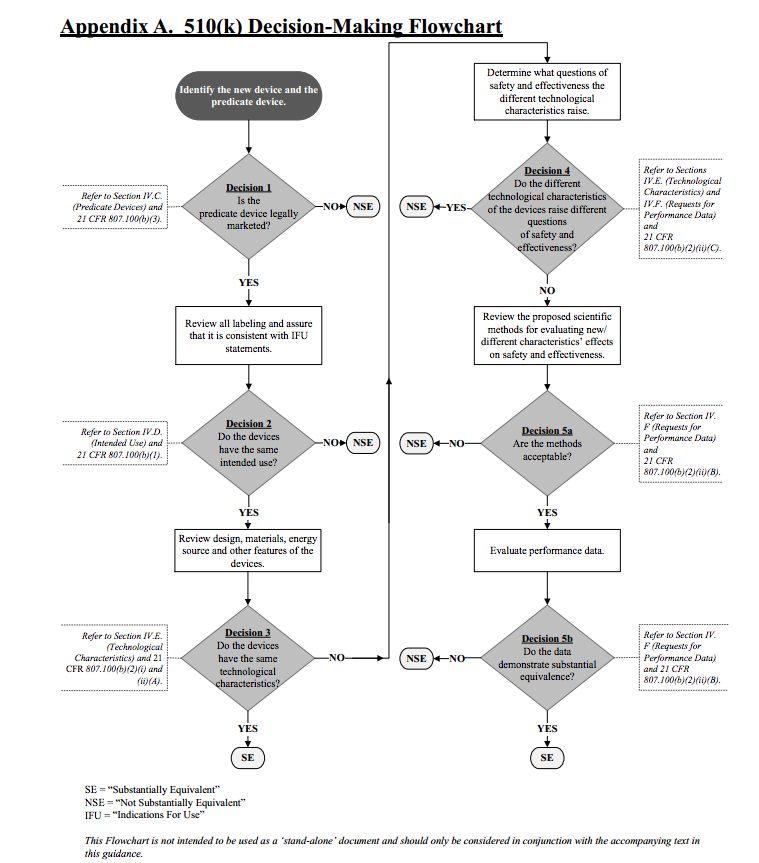
Nachweis der Äquivalenz
Damit der Hersteller dieses Predicate Device anführen darf, muss er nachweisen, dass es im Wesentlichen äquivalent ist („substantially equivalent“).
In den meisten Fällen möchten Hersteller aber kein identisches Produkt vermarkten (z. B. die x-te Spritze), sondern eines, das zwar die gleiche Zweckbestimmung hat, sich aber in der technischen Umsetzung unterscheidet.
Die überarbeitete Leitlinie Evaluating Substantial Equivalence in Premarket Notifications gibt Ihnen sehr konkrete Hinweise dazu, wann Sie ein Gerät als „substantially equivalent“ anführen und somit als Predicate Device für eine Premarket Notification referenzieren dürfen. Dabei untersucht die FDA folgende Aspekte:
- Haben die Produkte die gleiche Zweckbestimmung?
- Stellen sich keine neuen Fragen zur Sicherheit und Wirksamkeit?
- Sind die Produkte äquivalent (technisch, biologisch usw.)?
4. Kritik
Das Zulassungsverfahren beruht darauf, dass es ein Predicate Device gibt. Für dieses wurde bereit das Nutzen-Risiko-Verhältnis bewertet. Eine Neubewertung findet somit meist nicht mehr statt.
Allerdings kann dieses Predicate Device selbst wieder ein Predicate Device haben. So entstehen ganze Ketten von Referenzen, die letztlich bei Medizinprodukten enden, die vor Jahrzehnten erstmalig in den US-Markt kamen. Damit wird nicht mehr der aktuelle Stand der Technik gewährleistet.
Dies hat vor vielen Jahren bereits John Oliver kritisiert (s. Video).
Die FDA ist sich dieses Problems bewusst und arbeitet an neuen Zulassungsverfahren.
Im September 2023 hat die FDA erste Schritte zur Modernisierung des 510(k)-Verfahrens angestoßen und eine Reihe von Guidance-Dokumenten veröffentlicht. Beachten sollten Sie vor allem die Best Practices for Selecting a Predicate Device to Support a Premarket Notification [510(k)] Submission. Mit diesen „Best Practices“ stellt die FDA den Herstellern strengere Anforderungen bei der Auswahl eines Predicate Device. Demnach ist es nicht mehr möglich, jedes beliebige equivalente Produkt auszuwählen als Predicate. Hersteller müssen nun genauer recherchieren und nur solche Vergleichsprodukte auswählen, die möglichst nach aktuellen (anerkannten) Normen entwickelt und hergestellt wurden und im besten Fall keine Design-bedingte Rückruf-Historie aufweisen.
5. Fazit und Zusammenfassung
Noch ist das 510(k) das beliebteste Zulassungsverfahren in den USA. Es ist verständlich beschrieben und planbar, und wenn alle Unterlagen vorliegen, ist die FDA verpflichtet, innerhalb von 90 Tagen zu entscheiden.
Allerdings führt das dazu, dass Hersteller eher „me too“-Produkte zulassen als innovative Produkte. Daher wird das Zulassungsverfahren künftig an Bedeutung verlieren.
Beachten Sie auch unseren Beitrag Die fünf häufigsten Fehler bei 510k-Zulassungen vermeiden.
Die Regulatory Affairs Experten des Johner Instituts unterstützen bei der weltweiten Zulassung von Medizinprodukten.
Wir helfen Ihnen gerne, beispielsweise bei der regulatorischen Strategie für die USA, einer Predicate Device-Strategie oder der gesamten 510(k)-Einreichung. Bei Bedarf übernehmen wir auch die Rolle des US Agents.
Melden Sie sich, beispielsweise über das Kontaktformular.
Der Auditgarant erklärt schrittweise
- die Anforderungen der FDA,
- wie Predicate Devices gesucht und begründet werden,
- wie die Zulassungsdokumentation erstellt wird und
- was Hersteller bei „Registration“, „Listing“ und nach der Inverkehrbringung tun müssen.
Änderungshistorie
- 2023-10-24: Referenz auf neuen Artikel zum „Predicate Device“ und zur „Substantial Equivalence“ ergänzt
- 2023-09-14: Kapitel 2 überarbeitet (eSTAR-Format). Neueste Informationen in Kapitel 4 zur Modernisierung des 510(k)-Verfahrens ergänzt.


