
Die EN ISO 14155:2020 ist eine für die MDR bisher nicht harmonisierte Norm mit dem Titel „Clinical investigations of medical devices for human subjects – good clinical practice”. Auf Deutsch: „Klinische Prüfung von Medizinprodukten – Gute klinische Praxis“.
Sie beschreibt somit für Medizinproduktehersteller den Stand der Technik bei der Vorbereitung, Planung, Durchführung und Auswertung klinischer Prüfungen und bestimmt die Verantwortlichkeiten der beteiligten Akteure, insbesondere des sogenannten Sponsors.
Dieser Artikel hilft mit einer Übersicht über die ISO 14155 und mit Tipps zur Anwendung der Norm, die regulatorischen Anforderungen an klinische Prüfungen möglichst schnell und unaufwändig zu erfüllen.
1. ISO 14155: Eine Einführung
1.1 Kontext
Die MDR hat die Anforderungen an klinische Daten erhöht, mit denen Medizinproduktehersteller die Leistungsfähigkeit, Sicherheit und Wirksamkeit ihrer Produkte nachweisen müssen. Daher sind jetzt mehr Hersteller verpflichtet, diese Daten im Rahmen von klinischen Studien, insbesondere klinischen Prüfungen, zu erheben. An diese klinischen Prüfungen stellt die MDR ebenfalls umfangreiche Anforderungen.
Als für die MDR künftig harmonisierte Norm soll die ISO 14155 den Herstellern beim Nachweis dieser regulatorischen Anforderungen dienen.
1.2 Anwendungsbereich der ISO 14155
Die Norm ist zum einen anwendbar bei klinischen Prüfungen von Medizinprodukten, die dem Ziel dienen, die Leistungsfähigkeit, Sicherheit und Wirksamkeit der Produkte nachzuweisen. Zum anderen fallen auch die sonstigen „klinischen Untersuchungen nach dem Inverkehrbringen“ (so der Wortlaut der Norm) in den Anwendungsbereich der ISO 14155. Das sind beispielsweise Post-Market Clinical-Follow-up (PMCF)-Studien.
1.3 Ziele der Norm
Die Norm verfolgt mehrere Ziele:
- Schutz der Probanden (Sicherheit, Wohlbefinden, Rechte)
- Belastbarkeit der Ergebnisse
- Klarheit über die Verantwortlichkeiten, insbesondere des Sponsors (z. B. Herstellers), Hauptprüfers und der potenziellen weiteren Beteiligten (z. B. CRO, Krankenhaus)
- Einheitliches Verständnis bei allen Beteiligten (Sponsoren, Prüfer, Ethik-Kommissionen, Behörden, Benannte Stellen)

1.4 Nutzen für die Hersteller
Deshalb profitieren die Medizinproduktehersteller von der Norm:
- Weniger regulatorische Unsicherheit und Diskussion mit Behörden und Benannten Stellen
- Dadurch schnellere und weniger aufwendige „Zulassung“ der Produkte
- Zeitersparnis durch konkrete Vorgaben und Beispiele, z. B. zur Struktur von Dokumenten wie dem klinischen Prüfplan und dem klinischen Prüfbericht
2. Aufbau der ISO 14155:2020
Die Norm umfasst über hundert Seiten und besteht aus zehn Kapiteln und zehn Anhängen (s. Abb. 2).
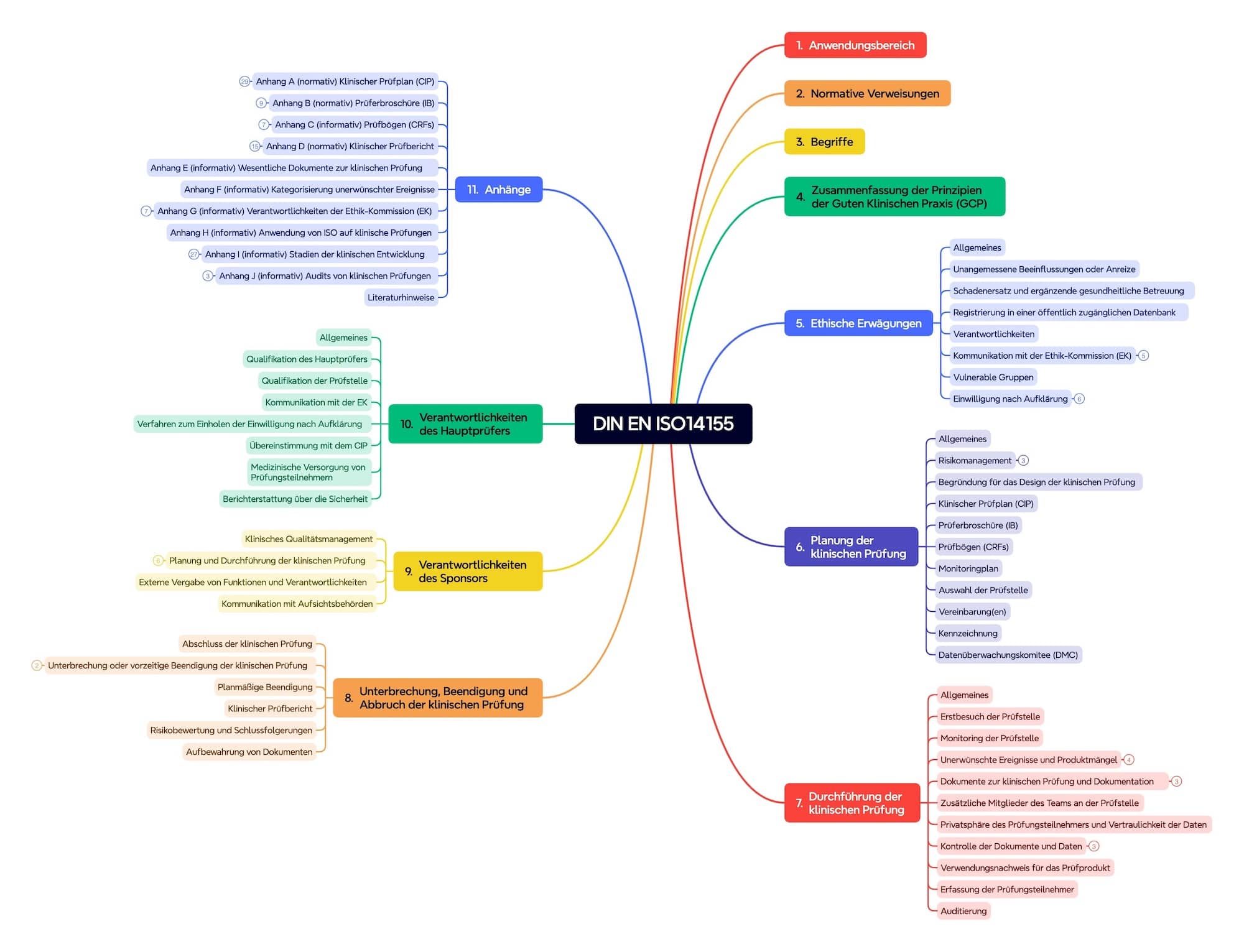
Kapitel 4: Gute klinische Praxis
Das Kapitel 4 fasst die Prinzipien der guten klinischen Praxis (GCP) zusammen.
Kapitel 5: Ethische Erwägungen
Das Kapitel 5 enthält die ethischen Erwägungen. Es betrachtet u. a. die Kommunikation mit der Ethik-Kommission, Verantwortlichkeiten, vulnerable Gruppen, Einwilligungserklärung und Patienteninformation.
Kapitel 6: Planung der klinischen Prüfung
Kapitel 6 beschreibt die Planung der klinischen Prüfung. Das beinhaltet:
- Vorgaben zum Prüfprodukt
- Vorgaben zum Design der klinischen Prüfung
- Vorgaben zur Auswahl der Prüfstelle und den essenziellen Dokumenten der klinischen Prüfung
Zu diesen Dokumenten zählen:
- Klinischer Prüfplan (CIP – Clinical Investigation Plan)
- Prüferbroschüre (IB – Investigators Brochure)
- Prüfbögen (CRFs – Case-Report-Forms)
Die Anhänge geben weitere Hinweise zum Aufbau dieser Dokumente.
Kapitel 7: Durchführung der klinischen Prüfung
Das Kapitel 7 benennt die Anforderungen an die Durchführung der klinischen Prüfung:
- Angaben zur praktischen Durchführung
- Meldungen von Ereignissen oder Produktmängeln
- Handling von Dokumenten und Daten
- Durchführung des Monitorings
- Vertraulichkeit der Daten und Identifizierung von Prüfungsteilnehmern
Kapitel 8: Unterbrechung, Beendigung und Abbruch der klinischen Prüfung
Hier sind die Vorgaben an die Unterbrechung, Beendigung und den Abbruch der klinischen Prüfung enthalten. Diese betreffen auch den klinischen Prüfbericht, die Risikobewertung und die Aufbewahrung der Dokumente.
Kapitel 9 und 10: Verantwortlichkeiten
Die Kapitel 9 und 10 regeln die Verantwortlichkeiten des Sponsors und des Hauptprüfers. Für Medizinproduktehersteller sind besonders die Anforderungen an den Sponsor relevant.
Normative und informative Anhänge
Die umfangreichen Anhänge der ISO 14155 sind teilweise normativ.
- Klinischer Prüfplan (CIP) (normativ): Festgelegte Inhalte des CIP
- Prüferbroschüre (IB) (normativ): Mindestanforderungen an die Inhalte der IB
- Prüfbögen (CRFs) (informativ): Tipps zur Erstellung von CRFs
- Klinischer Prüfbericht (normativ): Festgelegte Inhalte des klinischen Prüfberichts zum Abschluss der klinischen Prüfung
- Wesentliche Dokumente zur klinischen Prüfung (informativ): Dokumente, die vor, während und nach der klinischen Prüfung beim Sponsor oder der Prüfstelle zur Verfügung stehen müssen
- Kategorisierung unerwünschter Ereignisse (informativ): Übersichtstabelle und Grafiken zur Orientierung
- Verantwortlichkeiten der Ethik-Kommission (EK) (informativ): Leitfaden für die gute Praxis bei Ethik-Kommissionen
- Anwendung der ISO 14971 auf klinische Prüfungen (informativ): Grafik zum Umgang mit Risiken bei einer klinischen Prüfung
- Stadien der klinischen Entwicklung (informativ): Übersicht zu den verschiedenen entwicklungsbegleitenden Typen von klinischen Prüfungen des Medizinproduktes und Anwendbarkeit der ISO 14155 auf die verschiedenen Typen
- Audits von klinischen Prüfungen (informativ): Tipps, wie der Sponsor nachweisen kann, dass die klinische Prüfung in Einklang mit GCP erfolgt
3. Wichtige Anforderungen der ISO 14155
3.1 Ethische Überlegungen
Klinische Prüfungen müssen in Übereinstimmung mit ethischen Grundsätzen durchgeführt werden. Die ISO 14155 ist immer anzuwenden mit der aktuellen Version der Deklaration von Helsinki, in der die ethischen Grundsätze zur Durchführung von klinischen Prüfungen festgelegt sind.
Vorhersehbare Risiken müssen gegen den erwarteten Nutzen abgewogen werden.
Die Rechte, Sicherheit und das Wohl der Teilnehmer haben Priorität vor wissenschaftlichen Interessen.
3.2 Bedingungslose wissenschaftliche Rigorosität
Die Prüfung muss klaren und nachvollziehbaren statistischen Vorgaben folgen, die fundierte Ergebnisse mit hoher Evidenz bringen.
Präklinische und klinische Informationen müssen die Prüfung hinreichend stützen.
3.3 Umfassende Dokumentation
Es ist eine gründliche und nachvollziehbare Dokumentation aller Phasen der klinischen Prüfung erforderlich.
Die Durchführung muss gemäß den von Ethik-Kommission und Behörde genehmigten Dokumenten erfolgen (u. a. Prüfplan, Handbuch des klinischen Prüfers, Patienteninformation).
3.4 Klare Verantwortlichkeiten
Klare Definitionen der Rollen und Verantwortlichkeiten aller Beteiligten sind unerlässlich und müssen schriftlich festgelegt werden.
4. Stolpersteine, die es zu vermeiden gilt
4.1 Nicht erfüllte Voraussetzungen beim Hersteller
Oft herrscht die Annahme, der Hersteller könne ohne weiteres die Rolle des Sponsors in der klinischen Prüfung übernehmen.
Die Sponsorenschaft im Rahmen einer klinischen Studie ist mit einigen Vorgaben verbunden, u. a. mit der Integration eines klinischen Qualitätsmanagements in das bereits bestehende QMS des Herstellers.
Denken Sie daher frühzeitig daran, dass Sie die Prüfung erst beginnen können, wenn Sie als Sponsor die Anforderungen der Norm und der MDR erfüllen.
4.2 Irrtum über die Anwendbarkeit der ISO 14155
„Wir machen nur eine PMCF-Studie, das Produkt ist schon zertifiziert, da gilt die Norm nicht.“
Das ist eine Fehlannahme.
Die Norm gilt nicht nur für „Zulassungsstudien“, die zur Erlangung des CE durchgeführt werden. Sie ist in Teilen ebenfalls anzuwenden bei Studien zur Klärung von wissenschaftlichen Fragestellungen (Artikel 82 nach MDR) oder auch bei PMCF-Studien (Artikel 74 Studien). Dies wird im Anhang I.7 der Norm erläutert.
4.3 Fehlende Dokumentation
Zum Nachweis der klinischen Evidenz reicht die Zusammenfassung der Studienergebnisse in der Regel nicht aus.
Hersteller müssen die klinischen Daten des eigenen Produkts aus Studien im Rahmen der klinischen Bewertung evaluieren. Wenn Sie eine Studie mit Ihrem eigenen Produkt durchführen, wird erwartet, dass Sie zum Nachweis der klinischen Evidenz den Prüfplan der Studie vorlegen und die Konformität der Studiendurchführung mit der ISO 14155 belegen können. Dies wird im Rahmen des „Clinical Evaluation Assessment“ von der Benannten Stelle geprüft (Sektion E des MDCG 2020-13).
Im schlimmsten Fall könnten Zweifel an der Qualität der Daten entstehen, da ein Hersteller ohne den Prüfplan und die anderen Dokumente nicht nachweisen kann, dass die Studie z. B. nach ISO 14155 durchgeführt wurde. Das würde bedeuten, dass Sie ggf. die Daten ausschließen müssten.
4.4 Ausschließlicher Fokus auf ISO 14155
Es ist ein weit verbreiteter Irrtum, dass die reine Einhaltung der ISO 14155 automatisch alle nationalen Anforderungen erfüllt. Richtig ist hingegen, dass nationale Vorgaben, insbesondere außerhalb der EU, zusätzlich berücksichtigt werden müssen.
5. Versionen der ISO 14155
5.1 Unterschiede zwischen den Versionen 2012 und 2020
Die ISO 14155:2020-01 (bzw. DIN EN ISO 14155:2021-05) hat im Vergleich zur Vorgängernorm aus dem Jahr 2012 mehrere nennenswerte Änderungen umgesetzt:
- Die Verflechtung mit der ICH-GCP Guideline wurde stärker hervorgehoben und eine Zusammenfassung der GCP-Grundsätze wurde in Abschnitt 4 der Norm integriert.
- Das Risikomanagement wurde in den gesamten Prozess der klinischen Prüfung integriert.
- Der für Hersteller besonders interessante Anhang I wurde eingeführt, der die Anwendbarkeit der Norm auf die unterschiedlichen klinischen Entwicklungsstufen des Medizinprodukts erläutert.
- Eine Anforderung zur Registrierung der klinischen Prüfung in einer öffentlich zugänglichen Datenbank wurde ergänzt (neu in der Version 2020/2021).
- Das klinische Qualitätsmanagement (Abschnitt 9.1) und das risikobasierte Monitoring (Abschnitt 6.7) wurden integriert.
- Die Anpassung an internationale Datenschutzanforderungen wurde in der aktuellen Version berücksichtigt.
5.2 Ausblick auf die nächste Version
Derzeit wird an einer Änderung der der ISO 14155 gearbeitet. Der Veröffentlichungszeitpunkt ist im Moment noch unbekannt (Stand Juni 2025).
Die Überarbeitung zielt darauf ab, die klinischen Prüfungen mit den EU-Vorgaben der MDR zu harmonisieren. Sie soll ohne Übergangsfrist gültig werden. (Quelle)
6. Fazit und Zusammenfassung
Die ISO 14155 ist – unabhängig von deren Harmonisierung – der Goldstandard für die Vorbereitung, Planung, Durchführung und Auswertung klinischer Studien von Medizinprodukten. Sie ist nicht nur bei klinischen Prüfungen anwendbar und sollte auch bei Studien außerhalb Europas befolgt werden.
Die Norm ist hinreichend präzise und vollständig und daher den Herstellern eher ein Nutzen als eine weitere regulatorische Bürde.
Die „Clinical Experts“ des Johner Instituts helfen Ihnen, klinische Daten zu finden und zu entscheiden, ob eine klinische Studie überhaupt notwendig ist.
Falls dies der Fall ist, unterstützen wir Sie bei Ihrer kompletten klinischen Prüfung oder PMCF-Studie: von der Planung über die Beantragung bis zur Durchführung.
- Studiendesign: Wir helfen beim Design und der Abstimmung.
- Statistische Fallzahlplanung: Optimale Planung für valide Ergebnisse
- CRO-Auswahl: Wir finden die passende Contract Research Organization (CRO).
- Einreichung bei Behörden: Erstellung regulatorisch konformer Unterlagen
- Durchführung und Auswertung: Stets an Ihrer Seite während der Zusammenarbeit mit der CRO im klinischen Feld
Sie finden hier eine Übersicht über die Unterstützung des Johner Instituts bei klinischen Bewertungen und Prüfungen.


Ich finde es missverständlich, bei der Klärung von allgemeinen wissenschaftlichen Fragestellungen auf Artikel 82 MDR zu verweisen, da sich Artikel 82 auch den Begriff „clinical investigation“ verwendet, bei der es immer um die Untersuchung der EIgenschaften von Medizinprodukten geht.
[MDR Art. 2 (45) ‘clinical investigation’ means any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device;]
Sehr geehrter Herr Müller,
Sie haben recht, das Wording in der MDR ist missverständlich! Allerdings schreibt die MDR in Artikel 82 (2) auch: „legt jeder betroffene Mitgliedstaat für ihn geeignete zusätzliche Anforderungen für diese Prüfungen fest“. In Deutschland wurde im Rahmen des MPDG folgendes in §3 Abschnitt 4 definiert:
„„sonstige klinische Prüfung“ eines Produktes eine klinische Prüfung, die
a) nicht Teil eines systematischen und geplanten Prozesses zur Produktentwicklung oder der Produktbeobachtung eines gegenwärtigen oder künftigen Herstellers ist,
b) nicht mit dem Ziel durchgeführt wird, die Konformität eines Produktes mit den Anforderungen der Verordnung (EU) 2017/745 nachzuweisen,
c) der Beantwortung wissenschaftlicher oder anderer Fragestellungen dient und
d) außerhalb eines klinischen Entwicklungsplans nach Anhang XIV Teil A Ziffer 1 Buchstabe a der Verordnung (EU) 2017/745 erfolgt;“
In Verbindung mit der ergänzenden Definition des MPDG lässt es sich etwas besser nachvollziehen.
Viele Grüße,
Susanne Golombek