Die Qualifizierung und Klassifizierung sind Einteilungen in „Klassen“, mit denen Hersteller, Benannte Stellen und Behörden die regulatorischen Anforderungen an die Medizinprodukte bestimmen.
Inhalt
Diese Seite verschafft einen Überblick und verlinkt auf relevante Fachartikel:
- Fachartikel zur Qualifizierung und Klassifizierung
- Weiterführende Artikel zu diesem Thema
- Unterstützung bei der Qualifizierung und Klassifizierung
1. Fachartikel zur Qualifizierung und Klassifizierung
a) Übersicht über die Einstufung in Klassen
Vorsicht!
Der Begriff „Klassifizierung“ ist bei Medizinprodukten mehrfach belegt. Zudem wird häufig von einer Klassifizierung gesprochen, wenn eine Qualifizierung gemeint ist.
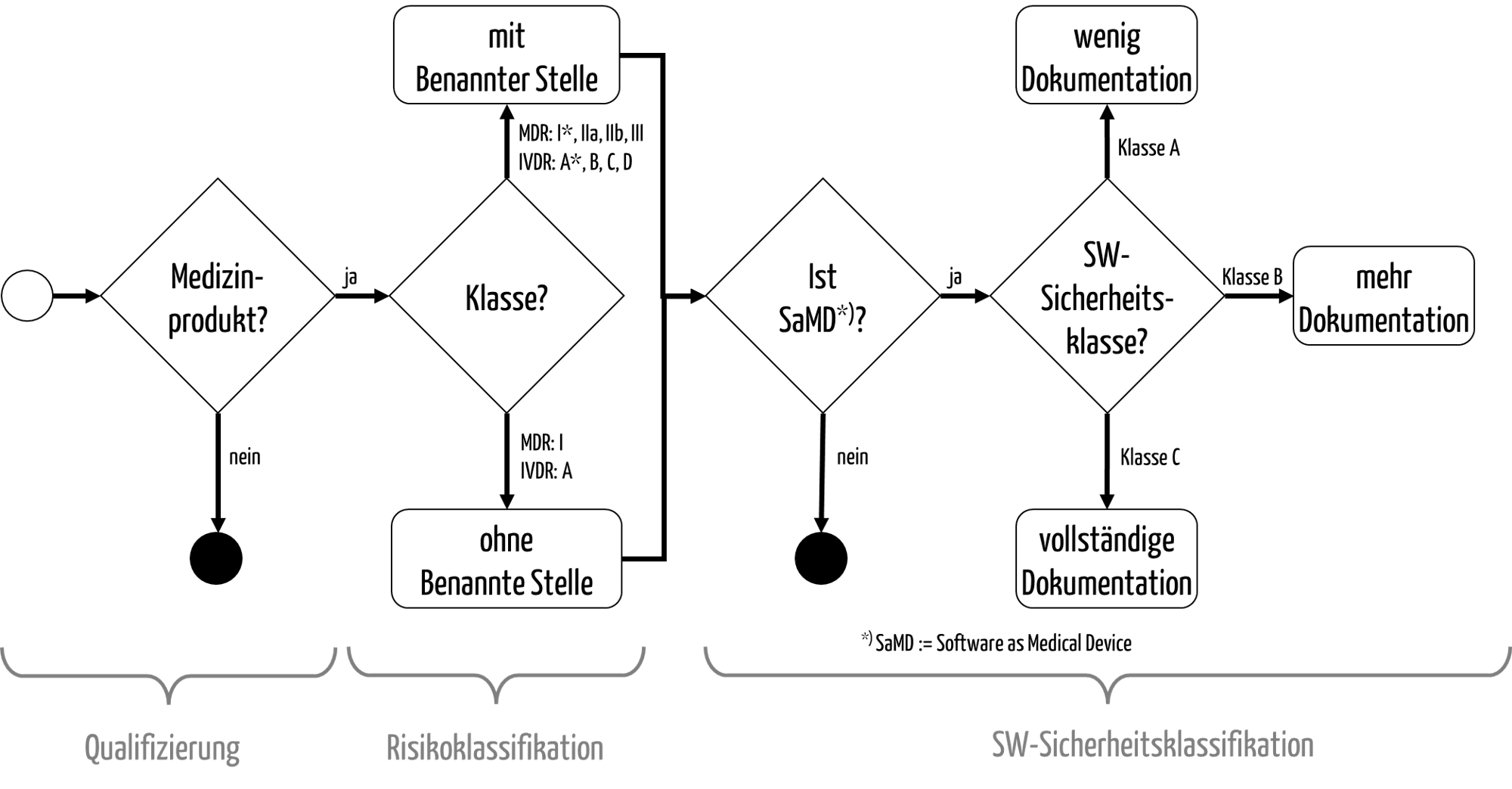
Abb. 1: Die Qualifizierung und Klassifizierung von Medizinprodukten
b) Qualifizierung: Was sind Medizinprodukte?
Die erste Entscheidung, die Einstufung als Medizinprodukt (oder eben nicht), ist die Qualifizierung. Dabei wird entschieden, ob ein Produkt ein Medizinprodukt (oder ein Zubehör) ist oder nicht.
Die EU-Verordnungen MDR und IVDR qualifizieren, vergleichbar der FDA – vereinfacht ausgedrückt – alle Produkte als Medizinprodukte, die vom Hersteller dazu bestimmt sind, Krankheiten und Verletzungen von Patienten zu diagnostizieren, zu überwachen, zu lindern und zu therapieren.
Die genaue Definition findet sich in der MDR im Artikel 2, Absatz 1.
„Medizinprodukt“ bezeichnet ein Instrument, einen Apparat, ein Gerät, eine Software, ein Implantat, ein Reagenz, ein Material oder einen anderen Gegenstand, das dem Hersteller zufolge für Menschen bestimmt ist und allein oder in Kombination einen oder mehrere der folgenden spezifischen medizinischen Zwecke erfüllen soll:
- Diagnose, Verhütung, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten,
- Diagnose, Überwachung, Behandlung, Linderung von oder Kompensierung von Verletzungen oder Behinderungen,
- Untersuchung, Ersatz oder Veränderung der Anatomie oder eines physiologischen oder pathologischen Vorgangs oder Zustands,
- Gewinnung von Informationen durch die In-vitro-Untersuchung von aus dem menschlichen Körper — auch aus Organ-, Blut- und Gewebespenden — stammenden Proben
und dessen bestimmungsgemäße Hauptwirkung im oder am menschlichen Körper weder durch pharmakologische oder immunologische Mittel noch metabolisch erreicht wird, dessen Wirkungsweise aber durch solche Mittel unterstützt werden kann.
Die folgenden Produkte gelten ebenfalls als Medizinprodukte:
- Produkte zur Empfängnisverhütung oder -förderung,
- Produkte, die speziell für die Reinigung, Desinfektion oder Sterilisation der in Artikel 1 Absatz 4 genannten Produkte und der in Absatz 1 dieses Spiegelstrichs genannten Produkte bestimmt sind.
MDR Artikel 2 (1)
Diese Entscheidung über die Einstufung erfolgt fast immer anhand der Zweckbestimmung. Bei Software spielt laut MDCG 2019-11 auch die Funktionalität eine Rolle.
Beispiele für Medizinprodukte
Medizinprodukte sind u. a.:
- Medizinisch-elektrische Geräte wie Röntgengeräte, CTs, Kernspingeräte, Ultraschallgeräte, elektrische Blutdruckmessgeräte
- Chirurgische Instrumente, z. B. Skalpelle, Zangen, Pinzetten
- Hüftimplantate
- Viele Heil- und Hilfsmittel, z. B. Rollstühle und Verbandsmaterial
- Einige Software-Anwendungen, wie DiGA, PDMS und RIS
Beispiele für Produkte, die nicht als Medizinprodukte zählen
Nicht als Medizinprodukt zählen u. a.:
- Krankenhaus-Informationssysteme, die nur der Dokumentation dienen
- Die meisten Wellness- und Fitness-Produkte
- Arzneimittel
- Produkte, die vom Hersteller nicht speziell für die Anwendung am Patienten in den Markt gebracht wurden, aber dafür genutzt werden können
Ein Teil dieser Qualifizierung sind auch die Einteilungen:
Sonderfall: Medizinprodukte gemäß Anhang XVI MDR
Einen Sonderfall bilden die Produkt ohne medizinische Zweckbestimmung, die dennoch den Anforderungen der MDR genügen müssen.
Beispiele sind „Schönheitslaser“ oder Produkte zur Fettabsaugung. Welche Produkte zu diesen Sonderfälle zählen, legt die MDR im Anhang XVI fest.
Beachten Sie den Fachartikel zu den Produkten ohne medizinische Zweckbestimmung und zum Anhang XVI.
Vorsicht!
Bei vielen Produkten ist es eine wichtige strategische Entscheidung, welche Teile eines Systems der Hersteller als ein oder mehrere Medizinprodukte bzw. als Zubehör oder Nicht-Medizinprodukt in den Verkehr bringt. Das Johner Institut hilft dabei (siehe unten).
c) Klassifizierung der Medizinprodukte (Einstufung der „Risikoklassen“)
Übersicht
Die anschließende Klassifizierung dient dazu, die möglichen Konformitätsbewertungsverfahren bzw. Zulassungsverfahren zu bestimmen (s. Abb. 1).
- Die MDR unterscheidet die Klassen I, I*, IIa, IIb und III.
- Bei der IVDR gibt es die Unterscheidung in die Klassen A, B, C und D.
- Die FDA unterteilt die Produkte in die Klassen I, II und III.
Nur für die Produkte der niedrigsten „Risikoklasse“ muss in Europa keine Benannte Stelle in das Konformitätsbewertungsverfahren einbezogen werden.
Risikoklassifizierung in der EU
Die EU unterscheidet bei der Einteilung in Risikoklassen auch folgende Produktklassen:
- Medizinprodukte mit oder ohne Messfunktion
- Medizinprodukte, das als wiederverwendbares chirurgisches Instrument zählt
- Sterile Medizinprodukte
Produkte, die in eine dieser Klassen fallen und sonst der Klasse I zugeordnet wären, zählen zur Klasse I*.
Weiterführende Informationen
Zu besonders vielen Diskussionen führt in der EU die Regel 11 bei Medizinprodukten, die Software enthalten oder Software sind.
Weitere Hilfestellung bei der Einteilung in Risikoklassen gibt die Leitlinie MDCG 2021-24. Darin erläutern die Autoren die Klassifizierungsregeln der MDR und gehen dabei beispielsweise darauf ein, was ein implantierbares Medizinprodukt ist.
Zudem visualisiert die MDCG 2021-24 den Ablauf der Klassifizierung mit Hilfe von Entscheidungsbäumen.
Schließlich enthält die Leitlinie Beispiele für die Einstufung der Medizinprodukte in Risikoklassen.
Vorsicht!
Die Klassifizierung der Medizinprodukte ist trotz dieser Hilfsmittel Gegenstand von Streitigkeiten zwischen Hersteller einerseits und Benannten Stellen und Behörden wie dem BfArM andererseits. Das Johner Institut unterstützt die Hersteller bei der rechtskonformen Klassifizierung und Argumentation.
c) Software-Sicherheitsklassifizierung
Schließlich gibt es noch eine Klassifizierung speziell von Software, sei es „Software as a Medical Device“ (Standalone-Software) oder Software als Teil eines Medizinprodukts:
- Die IEC 62304 unterscheidet die Sicherheitsklassen A, B und C. Diese Klassen wirken sich auf den Umfang der zu erstellenden Dokumentation der Software aus (s. Abb. 1).
- Hingegen bestimmen die Levels of Concerns (jetzt „Documentation Levels“) die Art der einzureichenden Dokumentation der Software.
2. Weiterführende Artikel
Viele weitere Artikel sind bei der Qualifizierung und Klassifizierung von Medizinprodukten relevant:
Übergeordnete Überlegungen zur Einteilung von Konzepten stellt der Artikel „8 Tipps für präzises Schubladendenken“ an.
3. Unterstützung bei der Qualifizierung und Klassifizierung
Haben Sie noch Fragen zur Qualifizierung oder Klassifizierung von Medizinprodukten? Dann nutzen Sie das kostenfreie Micro-Consulting.
Die Experten und Expertinnen des Johner Instituts sind darauf spezialisiert, eine regulatorische Strategie zu erarbeiten, mit der Sie als Hersteller Ihre Produkte möglichst schnell und möglichst flexibel weltweit in den Markt bringen können. Melden Sie sich gleich hier.





