
Unter Software als Medizinprodukt (Software as Medical Device, SaMD) versteht man (eigenständige) Standalone-Software, die ein Medizinprodukt ist, aber nicht Teil eines solchen.
Sie ist nicht zu verwechseln mit Medical Device Software im Sinne der EU.
Wann müssen Sie als Hersteller Software als Medizinprodukt und wann als Medical Device Software qualifizieren? Das erfahren Sie hier – und können mit diesem Wissen juristische Konsequenzen und unnötige Aufwände vermeiden.
Die Entscheidung, ob eine Software ein Medizinprodukt ist, bezeichnet man als Qualifizierung. Umgangssprachlich wird oft von Klassifizierung gesprochen. Unter Klassifizierung versteht man aber die „Risikoklassifizierung“. Das ist die Einteilung in die Klassen, die die Regularien definieren, um die Zulassungsverfahren/Konformitätsbewertungsverfahren zu bestimmen. So kennt die MDR die Klassen I, IIa, IIb und III.
A) Definitionen
1. Software im medizinischen Kontext
Bei Software im Umfeld der Medizinprodukte unterscheidet man:
- Software als Teil eines Medizinprodukts, z. B. als embedded Software eines Medizingeräts
- Software als eigenständiges Medizinprodukt (Standalone-Software)
- Software, die ein Zubehör zu einem Medizinprodukt ist
- (Eigenständige) Software, die kein Medizinprodukt ist.
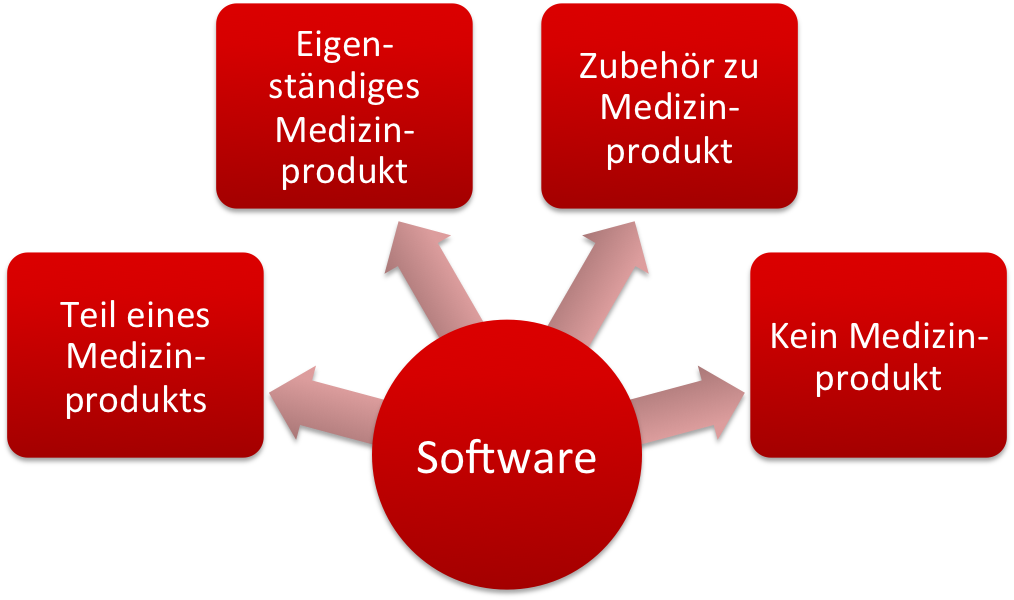
Abhängig von dieser Klassifizierung müssen Sie als Hersteller unterschiedliche Regularien beachten. Dieser Artikel hilft Ihnen, Ihre Software richtig zu qualifizieren.
Der Kategorieartikel „Regulatory Affairs“ verschafft eine Übersicht über alle regulatorischen Anforderungen an Medizinprodukte und IVF.
2. Definitionen
Es existieren mehrere Definitionen dessen, was unter Software als Medizinprodukt (Software as Medical Device, SaMD) zu verstehen ist. Eine Definition stammt vom IMDRF, dem International Medical Device Regulators Forum.
„software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device“
Quelle: IMDRF/SaMD WG/N12FINAL:2014
Nach Auskunft des deutschen Bundesgesundheitsministeriums ist im Leitfaden zur MDR folgende Definition des Begriffs „medizinische Software“ geplant, wie sie von der MDCG erarbeitet wird:
„Medical device software is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a ‚medical device‘ in the medical devices regulation or in vitro diagnostic medical devices regulation.“
Quelle: MDCG 2019-11 „Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR“
Zusätzlich zu der Definition schreibt die MDCG:
„Medical device software is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a ‚medical device‘ in the MDR or IVDR, regardless of whether the software is independent or driving or influencing the use of a device.“
Quelle: MDCG 2019-11
Damit umfasst Medical Device Software sowohl die sogenannte independent software (also die Standalone-Software) als auch die Steuerungssoftware (oft auch als Funktions-Software bezeichnet).
Diese Definition und das Guidance-Dokument der MDCG haben starke Auswirkungen auf die Klassifizierung von Software und die Anwendbarkeit der Regel 11.
Lesen Sie hier mehr zum Thema MDCG und Regel 11.
Das IMDRF gibt weitere „Key Definitions“ im SaMD-Kontext im Dokument IMDRF/SaMD WG/N10Final:2013.
B) Qualifizierung (Klassifizierung) von Software als Medizinprodukt
Die Entscheidung darüber, ob eine Software als Medizinprodukt zählt, bezeichnet man als Qualifizierung, auch wenn umgangssprachlich oft von Klassifizierung gesprochen wird.
Die Zweckbestimmung des Herstellers entscheidet …
Die Frage, wann Software als Medizinprodukt zu qualifizieren ist, stellt sich nur bei Standalone-Software. Die Antwort lautet in Kurzform: Software ist ein Medizinprodukt, wenn der Hersteller sie zur Diagnose, Therapie oder Überwachung von Krankheiten und Verletzungen vorgesehen hat. Punkt.
Exakter und formaler: Software ist genau dann ein Medizinprodukt, wenn die Zweckbestimmung des Herstellers der Definition des Begriffs „Medizinprodukt“ gemäß Artikel 2 (1) der MDR entspricht.
… und nicht primär die Funktionen der Software
Entscheidend bei der Qualifizierung ist also die Zweckbestimmung durch den Hersteller und weniger die Funktion der Software. Bei einer Software, die Vitalparameter erfasst, ließe sich diesbezüglich auf zwei Weisen argumentieren, die zu unterschiedlichen Bewertungen führen:
- Der Hersteller sagt: Die Erfassung dient nur der Dokumentation.
Dann ist das Produkt kein Medizinprodukt. - Der Hersteller sagt: Der Arzt kann anhand dieser Vitalparameter (und ggf. deren grafischer Darstellung) Trends erkennen und damit das richtige Medikament auswählen.
Dann ist das identische Produkt ein Medizinprodukt.
Und wenn es keine Zweckbestimmung gibt?
Wenn der Hersteller die Zweckbestimmung nicht eindeutig und widerspruchsfrei formuliert (Handbuch, Verpackung, Webseite, Marketingmaterialien usw.) und sein Produkt nicht als Medizinprodukt klassifiziert, kann es passieren, dass die „liebe Konkurrenz“ eine Abmahnung verschickt. Falls das bei Ihnen der Fall sein sollte, dann melden Sie sich. Wir kennen das.
Falls die Sache vor Gericht landet, wird der Richter wahrscheinlich einen Gutachter beauftragen. Dieser wiederum wird versuchen, aus den Unterlagen (Handbuch etc.) und aus den Funktionen(!) den vom Hersteller vorgesehenen Gebrauch (Zweckbestimmung) abzuleiten. Ob die Schlussfolgerungen des Gutachters in Ihrem Sinne sind, darf bezweifelt werden.
Wer über die Qualifizierung/Klassifizierung von Software als Medizinprodukt entscheidet
Häufig fühlen sich Hersteller und Behörden trotz der Definition des Begriffs unsicher, ob eine Software als Medizinprodukt einzustufen ist. Auch deshalb sind viele Entscheidungshilfen publiziert worden, die Ihnen dieser Artikel im Folgenden vorstellt.
Die Entscheidung, ob eine Software als Medizinprodukt zu qualifizieren ist, trifft der Hersteller selbst. Benannte Stellen können Gutachten erstellen. Damit erlangt der Hersteller aber keine rechtsverbindliche Auskunft. Anfragen beim BfArM werden in der Regel nicht zeitnah beantwortet. Letztlich, das mag zynisch klingen, wird erst ein Richter im Fall einer Klage die Frage nach der Qualifizierung/Klassifizierung final beantworten.
Wenn Sie unsicher sind, ob Ihre Software ein Medizinprodukt ist, dann wenden Sie sich
- an das Bundesinstitut für Arzneimittel und Medizinprodukte BfArM (Die Reaktionszeit ist aber nicht immer schnell.)
- an Benannte Stellen. Hier können wir Ihnen Kontakte verschaffen.
- an das Johner Institut. Wir beantworten ständig solche Fragen und erstellen Gutachten. Schnell und kompetent.
Sind Sie unsicher, ob Ihre medizinische Software ein Medizinprodukt ist? Benötigen Sie ein Gutachten? Professor Johner und sein Team helfen gerne. Nehmen Sie Kontakt auf!
Kontakt aufnehmen
C) Entscheidungshilfen zur Qualifizierung / Klassifizierung von Software as Medical Device SaMD
Das Thema „Klassifizierung von Software als Medizinprodukt“ beschäftigt nicht nur Medizinproduktehersteller, sondern auch Behörden, Gremien und Verbände. Diese haben eine Reihe von Dokumenten veröffentlicht, die als Entscheidungshilfen dienen sollen. Wir stellen Ihnen folgende Dokumente vor:
- COICR Contribution
- EU: Manual on Borderline and Classification
- UK Medicines and Healthcare Products Regulatory Agency
- International Medical Device Regulator Forum IMDRF
- MEDDEV 2.1.6
- Schwedische Behörden
- Asian Harmonization Working Group
- SwissMedic
- Britische MHRA
- FDA
- Health Canada
- MDCG (in Europa besonders relevant)
- Australisches Gesundheitsministerium
1. COICR Contribution
Das „European Coordination Committee of the Radiological, Electromedical and Healthcare IT Industry“ hat ein „Decision Diagram for Qualification of Software as Medical Device“ publiziert.
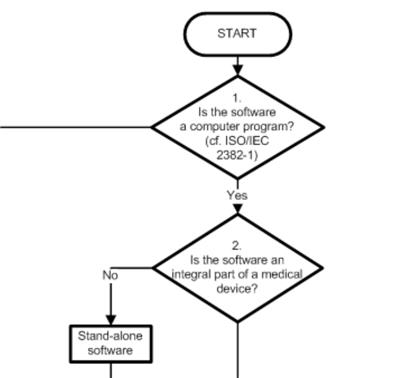
Über den Wert dieses Dokuments lässt sich streiten. Einerseits steht nichts wirklich Neues drin (und letztlich zählt nur die gesetzliche Definition). Andererseits haben sich hier Menschen viel Arbeit gemacht, um Hersteller und Entwickler zu unterstützen.
2. EU: Manual on Borderline and Classification
Von der EU stammt das Manual (V 1.22 (05-2019)), das anhand von Beispielen versucht, Medizinprodukte von Nicht-Medizinprodukten zu unterscheiden und Hilfe bei der Qualifizierung und Klassifizierung zu geben. Die Beispiele beziehen sich teilweise auch auf Software:
- Picture archiving and communication systems
- Mobile application for processing ECGs
- Mobile application for the communication between patient and caregivers while giving birth
- Mobile medical application for viewing the anatomy of the human body
Die EU hat eine Infografik publiziert, welche bei der Entscheidung helfen soll, ob eine Software als Medical Device Software (nicht SaMD!) zählt.
3. Medicines and Healthcare Products Regulatory Agency
Die britische „Medicines and Healthcare Products Regulatory Agency“ hat ein Dokument mit dem Titel „Borderlines with medical devices“ herausgegeben. Lesen Sie mal im Kapitel 9 den Satz zu den „Telecare Alarm Systems“. Ich wäre an Ihrer Einschätzung interessiert.
4. IMDRF
Zu den sehr relevanten Dokumenten zählt dieses Dokument des International Medical Device Regulator Forum (IMDRF). Es definiert folgende Begriffe:
- Software as a Medical Device (SaMD)
- Medical Device
- In Vitro Diagnostic Medical Device
- SaMD Changes
- SaMD Manufacturer
- Intended Use/Intended Purpose
Das Dokument ist mit zehn Seiten angenehm kurz und allemal einen Blick wert. Interessant sind die Referenzen, u. a. die Quellen:
- GHTF/SG1/N55:2008 Definition of the Terms Manufacturer, Authorised Representative,
Distributor and Importer - GHTF/SG1/N70:2011 Label and Instructions for Use for Medical Devices
- GHTF/SG1/N71:2012 Definition of Terms Medical Device and In Vitro Diagnostic
Medical Device - ISO/IEC 14764:2006 Software Engineering — Software Life Cycle Processes —
Maintenance
Das IMDRF geht nochmals auf die Definition des Begriffs Medizinprodukt (Software as Medical Device, SaMD) ein und nennt Fälle, unter denen eine Standalone-Software nicht dazu zählt. Umgekehrt gibt das Dokument Hinweise, wann diese Software der Definition entspricht, z. B.
- mitigation of a disease,
- provide information for determining compatibility, detecting, diagnosing, monitoring
- or treating physiological conditions, states of health, illnesses or congenital deformities,
- aid in diagnosis, screening, monitoring, predisposition, prognosis, prediction, determination of physiological status,
- aids for persons with disabilities.
Ob das wirklich bahnbrechende Erkenntnisse zur Qualifizierung von Software als Medizinprodukt sind, lässt sich sicher trefflich diskutieren. Schließlich geht die Hilfestellung nicht entscheidend über die Definition des Begriffs Medizinprodukt hinaus.
5. MEDDEV 2.1.6
Beachten Sie, dass das weiter unten diskutierte MDCG-Dokument 2019-11 die MEDDEV 2.1/6 für Software, die durch die MDR reguliert wird, obsolet macht!
Die MEDDEV 2.1/6 hatte im Juli 2016 eine aktuelle Version des Guidance on the Qualification and Classification of Stand Alone Software used in Healthcare within the Regulatory Framework of Medical Devices veröffentlicht. Man teilte Software in folgende Kategorien:
- Software, die kein Medizinprodukt ist, wie Entwicklungswerkzeuge oder Dokumentationssysteme, zu denen auch die KIS zählen (aber nur, solange sie nicht der Diagnose oder Therapie dienen)
- Software als Bestandteil eines Medizinprodukts, also Gerätesoftware
- Software, die selbst ein Medizinprodukt ist
- Software, die ein Zubehör darstellt
Ein im Vergleich zur Version von 2012 überarbeiteter Entscheidungsbaum sollte helfen, die richtige Klasse zu bestimmen:
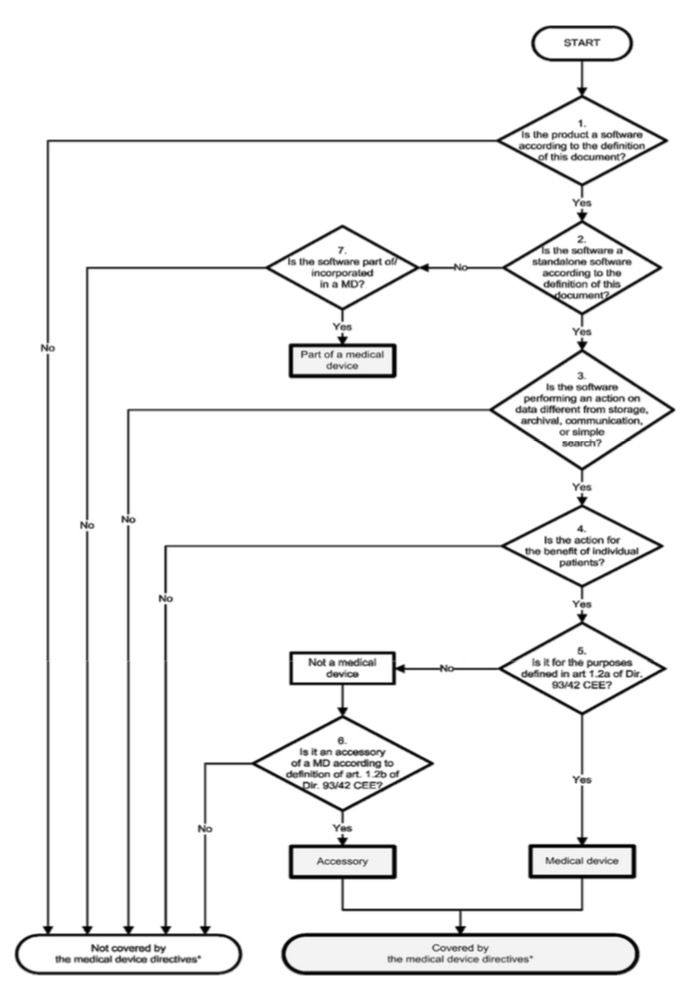
Dieses Dokument dürfte zu den wichtigsten in diesem Kontext gezählt haben.
Diese EU-Leitlinie gab auch Hilfestellung bei der Klassifizierung von IVD-Software.
Lesen Sie hier mehr zum Thema Klassifizierung von IVD-Software.
6. Schwedische Behörden
Von den schwedischen Behörden gab es den Leitfaden „Medical Information Systems – guidance for qualification and classification of standalone software with a medical purpose“. Dieser Link ist seit Februar 2020 nicht mehr aufrufbar.
7. Asian Harmonization Working Group — Software Qualification and Classification
Das Dokument der Asian Harmonization Working Group wiederholt die Definitionen im Kontext „Software als Medizinprodukt“ wie Standalone-Software, „Mobile Medical Application“ und „Health Software“. Es unterscheidet drei Typen:
- Software, die Teil eines Medizinprodukts ist (oft als embedded Software bezeichnet)
- (Standalone-) Software, die ein Zubehör für ein Medizinprodukt ist, z. B. zum Weiterleiten von Daten
- Standalone-Software, die selbst ein Medizinprodukt ist (Software as a Medical Device, SaMD) und auf Datenträger, per Download oder webbasiert zur Verfügung gestellt wird
Weiterhin stellt das Dokument bekannte Überlegungen zur Klassifizierung von Software als Medizinprodukt vor und diskutiert dabei im Wesentlichen die bereits oben genannten Quellen:
- Die Veröffentlichung des IMDRF (s. o.)
- Die australische Therapeutic Goods Association (TGA) referenziert selbst wieder europäische und US-amerikanische Veröffentlichungen. Sie klassifiziert Dokumentations-Software nicht als Medizinprodukt, ebenso wenig Software, die nur einfache Berechnungen ausführt, die nicht auf patientenspezifischen Daten basieren. Falls die Software ein weiteres Medizinprodukt kontrolliert oder beeinflusst, fällt es in die gleiche Klasse wie das Medizinprodukt.
- Die Chinese Food and Drug Administration CFDA hat eine genaue Richtlinie zur Registrierung von Software als Medizinprodukt veröffentlicht, die wiederum die Sicherheitsklassifizierung gemäß IEC 62304 verwendet.
- Von den in Europa veröffentlichten Dokumenten wird die MEDDEV 2.1/6 (s.o.) genannt, ebenso tabellarisch einige Beispiele für Software, die als Medizinprodukt oder als Nicht-Medizinprodukt zu klassifizieren wären. Auch werden die Klassifizierungsregeln des Anhangs IX der MDD zitiert.
- Kurz bespricht das Dokument die Vorgaben von Health Canada und erwähnt dabei v. a. ein FAQ-Dokument, das Beispiele und Regeln zur Klassifizierung von Software als Medizinprodukt gibt. Eine Tabelle nennt konkrete Beispiele.
- In den Vorgaben des japanischen MHLW (Ministry of Health, Labor and Welfare) steht, dass Standalone-Software ein Medizinprodukt sein kann und weitere Empfehlungen gerade erarbeitet werden.
- Eine Übersicht über die Vorgaben der FDA (s. o.) schließt den Reigen der untersuchten Rechtsbereiche ab.
In der Zusammenfassung stellt das Dokument Gemeinsamkeiten und Unterschiede u. a. tabellarisch zusammen. Dabei kommt auch Software zur Sprache, die der Datenkommunikation dient. Diese Software ist in Europa nicht als Medizinprodukt zu klassifizieren, in den meisten anderen Rechtsbereichen schon.
8. SwissMedic
Auch die SwissMedic hatte sich im AW-Merkblatt „Eigenständige Medizinprodukte-Software“ zu dem Thema geäußert. Sie fasst dabei viele der o. g. Quellen zusammen, ohne neue Erkenntnisse zu liefern. Interessant ist die klare Aussage, dass für „betriebsintern“ (z. B. innerhalb von Krankenhäusern) hergestellte Software die gleichen Voraussetzungen erfüllt sein müssen. Die Schweizer Behörde schreibt sogar:
„Betriebsintern hergestellte Software wird per Definition zwar nicht in Verkehr gebracht, die Anwendung durch eine Fachperson wird jedoch dem erstmaligen Inverkehrbringen gleichgesetzt.“
Inzwischen ist dieses Dokument inzwischen veraltet und wurde durch das Merkblatt BW630_30_007 ersetzt. Dessen Abschnitt 5.2 besagt, dass in Gesundheitseinrichtungen hergestellte Medizinprodukte-Software die grundlegenden Sicherheits- und Leistungsanforderungen (GSLA) gemäss Anhang I EU-MDR resp. Anhang I EU-IVDR erfüllen muss, sofern sämtliche Voraussetzungen nach Art. 5 (5) EU-MDR bzw. Art. 5 (5) EU-IVDR erfüllt sind.
9. Die britische MHRA
Die britische Medical & Healthcare products Regulatory Agency (MHRA) hat im September 2022 ein Dokument veröffentlicht, mit dem sie erklären will, wann Software ein Medizinprodukt ist und wann nicht.
Die Regeln sind im Wesentlichen dieselben wie in der Leitlinie MDCG 2019-11.
Die MHRA hat sich dabei viel Mühe gemacht, die Vorgaben und Regeln auch visuell ansprechend und leicht verständlich zu formulieren. Leider wirft auch sie Zweckbestimmung (z. B. „Prevention of Disease“), Funktionen („Stores or transmits medical data“) und technische Charakterisierungen (z. B. „Database without internal language“) durcheinander, was der Klarheit schadet.
10. FDA zur Qualifizierung und Klassifizierung von Software als Medizinprodukt
a) Food, Drug, and Cosmetic Act (FD&C)
Der amerikanische Food, Drug and Cosmetic Act, kurz FD&C, hat im Sommer 2017 die Definition des Begriffs ‚Medizinprodukt‘ speziell für Software überarbeitet. Allerdings ist diese Beschreibung so kryptisch geworden, dass die FDA im Dezember 2017 ein Guidance-Dokument veröffentlichte, das ihre Interpretation des Gesetzes darlegt. Diese Darlegung bezieht sich zwar nur auf Decision-Support-Systeme, ist aber auf andere Standalone-Software sehr gut übertragbar.
Lesen Sie hier mehr zum FDA Guidance Document on Decision Support Systems.
Hilfreich ist das Dokument der AAMI zur agilen Entwicklung von Medical Device Software.
Im September 2019 hat die FDA das Guidance-Dokument Changes to Existing Medical Software Policies Resulting from Section 3060 of the 21st Century Cures Act veröffentlicht. Darin legt die Behörde ihre Sicht der Dinge nochmals ausführlich dar, nennt Beispiele und verweist auf weitere (z. T. im Folgenden genannte) Dokumente.
b) General Wellness: Policy for Low Risk Devices
Bereits Ende November 2016 wurde der Food, Drug & Cosmetic Act überarbeitet. Er definiert Software „for maintaining or encouraging a healthy lifestyle and is unrelated to the diagnosis, cure, mitigation, prevention, or treatment of a disease or condition“ nicht(!) mehr als Medizinprodukt.
Die FDA bestätigt in ihrem Guidance-Dokument General Wellness: Policy for Low Risk Devices, dass sie weder untersuchen wolle, ob solche „low risk general wellness products“ Medizinprodukte seien, noch, ob solche Produkte, die als Medizinprodukt klassifiziert werden, den regulatorischen Anforderungen genügen.
Voraussetzung dafür sind:
- Die Produkte dienen ausschließlich dem allgemeinen Wohlbefinden („Wellness“) wie in diesem Dokument definiert.
- Die Produkte stellen ein niedriges Risiko dar.
Die Hersteller dürfen keinen Bezug zu Krankheiten herstellen, die mit den Produkten diagnostiziert oder behandelt werden sollen. Vielmehr geht es beispielsweise um
- Gewichtsmanagement (aber nicht Behandlung von Fettleibigkeit!),
- körperliche Fitness,
- Stress-Management,
- Schlaf.
Allerdings gestattet die FDA auch Produkte mit Bezug zu bestimmten Krankheiten in dieser Kategorie, falls sie ggf. dabei helfen,
- Risiken bestimmter chronischer Erkrankungen zu minimieren,
- mit bestimmten chronischen Erkrankungen besser zu leben.
Das Guidance-Dokument nennt Beispiele und liefert einen kleinen Entscheidungsbaum.
Fazit: Mit diesem Dokument schafft die FDA Klarheit, wann Produkte – auch Software – von der Überwachung durch die FDA ausgeschlossen sind. So etwas wünscht man sich in Europa auch. Die Behörde nimmt zudem viel mehr Produkte von der Überwachung aus als die europäischen Behörden und Regulatoren.
c) Policy for Device Software Functions and Mobile Medical Applications
Bereits die aus dem Jahr 2016 stammende Guidance zu Mobile Medical Apps grenzte Apps ab, die
- kein Medizinprodukt sind,
- ein Medizinprodukt sind, aber von der FDA nicht überprüft werden,
- ein Medizinprodukt sind, das den FDA-Überprüfungen unterliegt.
Mit dem Update im Jahr 2019 hat die FDA den Scope auch auf sonstige Software ausgedehnt und spricht jetzt häufig von „Software Functions“. Mobile Apps sind nur noch ein Typ von Software, die über solche Funktionen verfügt.
Das heißt, dass Sie das Dokument auch dann zurate ziehen sollten, wenn Sie bei Software, die keine Mobile Medical App ist, entscheiden müssen, ob sie ein Medizinprodukt ist.
Mehr dazu lesen Sie im Artikel über Mobile Medical Apps.
d) FDASIA Report
Die FDA ist Mitherausgeberin des FDASIA Reports. FDASIA steht für §Food and Drug Adminstration Safety and Innovation Act§, der von der FDA verlangt, einen Bericht zu erstellen,
„that contains a proposed strategy and recommendations on an appropriate, risk-based regulatory framework pertaining to health information technology, including mobile medical applications, that promotes innovation, protects patient safety, and avoids regulatory duplication.“
Die FDA unterscheidet (wie im Guidance-Dokument zu den Mobile Medical Apps):
- Nicht-Medizinprodukte
- Medizinprodukte, bei denen die FDA die Einhaltung der gesetzlichen Forderungen (z. B. Zulassungsverfahren, Quality Systems Regulations) einfordert
- Medizinprodukte, bei denen sie es nicht tut
Diese Dreiteilung ist überraschend. Gesetze müssen also nicht immer eingehalten werden? Die FDA begründet diese Differenzierung mit einem „risk-based approach”:
„In applying a risk-based approach, FDA does not intend to focus its regulatory oversight on these products/functionalities, even if they meet the statutory definition of a medical device.“
Sie nennt dann auch konkrete Beispiele, bei denen sie diese Ausnahmen für gerechtfertigt hält:
- Evidence-based clinician order sets tailored for a particular condition, disease, or clinician preference;
- Drug-drug interaction and drug-allergy contraindication alerts to avert adverse drug events;
- Most drug dosing calculations;
- Drug formulary guidelines;
- Reminders for preventative care (e.g. mammography, colonoscopy, immunizations, etc.);
- Facilitation of access to treatment guidelines and other reference material that can provide information relevant to particular patients;
- Calculation of prediction rules and severity of illness assessments (e.g., APACHE score, AHRQ Pneumonia Severity Index, Charlson Index);
- Duplicate testing alerts;
- Suggestions for possible diagnoses based on patient-specific information retrieved from a patient’s EHR.
Es wird nicht ganz klar, auf welcher statistischen Analyse die Annahme beruht, dass die o. g. Produktklassen wenige Risiken bergen. Solche Clinical-Decision-Support-Systeme sind oft besonders tückisch, weil sich die Risiken nicht so offenbaren wie bei klassischen Medizingeräten wie Beatmungsgeräten.
Achtung! Durch die im nächsten Abschnitt erwähnte Änderung der Section 520 des Federal Food, Drug, and Cosmetic Act (21 U.S.C. 360j) muss die FDA einige Guidance-Dokumente umschreiben, insbesondere zu Medical Device Data Systems (MDDS) und Mobile Medical Apps.
Ein neues Guidance-Dokument nennt konkrete Beispiele, wann Software nicht mehr als Medizinprodukt zu klassifizieren ist:
- Software zur administrativen Unterstützung
- Laborinformationssysteme (solange es nur um eine administrative Unterstützung, um Speicherung, Transport und Umwandlung von Labordaten geht)
- Software, die einen gesunden Lebensstil unterstützen will, z. B. eine App, die Bewegung aufzeichnet und darstellt, oder eine App, die den Benutzer warnt, wenn er zu viel oder ungesund isst. Auch Tagebücher und soziale Spiele zählen zu den Beispielen.
- Elektronische Patientenakten, allerdings nur, wenn drei Bedingungen erfüllt sind:
- Sie werden durch oder unter der Aufsicht von Healthcare Professionals gepflegt.
- Sie unterliegen nicht dem Health IT Certification Program.
- Sie dienen nicht der Analyse von Aufzeichnungen (einschließlich Bildern) mit dem Zweck einer Diagnose, Behandlung, Linderung oder Überwachung.
- Software, die nur der Weiterleitung, Speicherung, Formatumwandlung und Anzeige von Labordaten und anderen Gerätedaten dient, solange keine Analyse dieser Daten erfolgt. Damit sind die Medical Device Data Systems keine Medizinprodukte mehr. Auch PACS zählen nicht mehr zu den Medizinprodukten.
Entsprechend muss die FDA die oben genannten Guidance-Dokumente überarbeiten.
e) Digital Health Policy Navigator
Die FDA gibt Unterstützung bei der Qualifizierung und Klassifizierung von „Device Software Functions“. Sie hat dazu einen Digital Health Policy Navigator entwickelt, der in sieben Schritten bei der Entscheidungsfindung hilft.
Mehr dazu erfahren Sie diesem Draft Guidance der FDA zu „Existing Medical Software Policies“.
11. Health Canada
Health Canada hat den Entwurf des „Guidance Document – Software as a Medical Device (SaMD)“ herausgegeben. Es stützt sich im Wesentlichen auf das oben erwähnte „Framework“ des IMDRF und übernimmt dessen Definitionen. Es ergänzt dessen Gedanken durch Beispiele.
a) Medizinischer Zweck
So nennt Health Canada Beispiele Beispiele für eine medizinische Zweckbestimmung:
- Akquirieren, Verarbeiten oder Analysieren von medizinischen Bildern
- Akquirieren, Verarbeiten oder Analysieren von Informationen, die von einem IVD stammen
- Akquirieren, Verarbeiten oder Analysieren von Informationen, Messungen oder Signalen, die von einem Monitoring- oder bildgebenden Gerät stammen
- Unterstützung oder Bereitstellen von Empfehlungen für Patienten oder medizinisches Personal zur Vorbeugung, Diagnose, Behandlung, Linderung von Krankheiten oder Verletzungen
Allerdings qualifiziert Health Canada nicht alle entscheidungsunterstützenden Systeme als Medizinprodukt.
b) Beispiele für Software, die kein Medizinprodukt ist
Health Canada hilft auch mit Beispielen für Software, die die Behörde nicht zu den Medizinprodukten zählt:
- Reine Kommunikationssysteme wie MDDS, Telefonie etc.
- Software mit rein administrativen Zwecken einschließlich Workflow, Patientenverwaltung, Scheduling
- Software, die zu einem gesünderen Lebensstil beiträgt, wie Wellness Apps
- Elektronische Patientenakten
- Software, die Zugang zu Literaturdaten verschafft
- Software, die vor Medikamentenwechselwirkungen warnt
- Software, die keine unmittelbaren Handlungen auslöst
- Software für einfache medizinische Berechnungen
c) Risikoklassifizierung
Bei der Risikoklassifizierung lehnt sich Health Canada ebenfalls an das IMDRF-Dokument an, erlaubt aber an manchen Stellen eine niedrigere Klassifizierung.
12. MDCG
a) Übersicht
Das Dokument MDCG 2019-11 regelt die Qualifizierung und Klassifizierung von Medical Device Software unter der MDR. Es löst damit die oben erwähnte MEDDEV 2.1/6 ab. Zentral für viele Hersteller ist das Klassifizierungsschema:
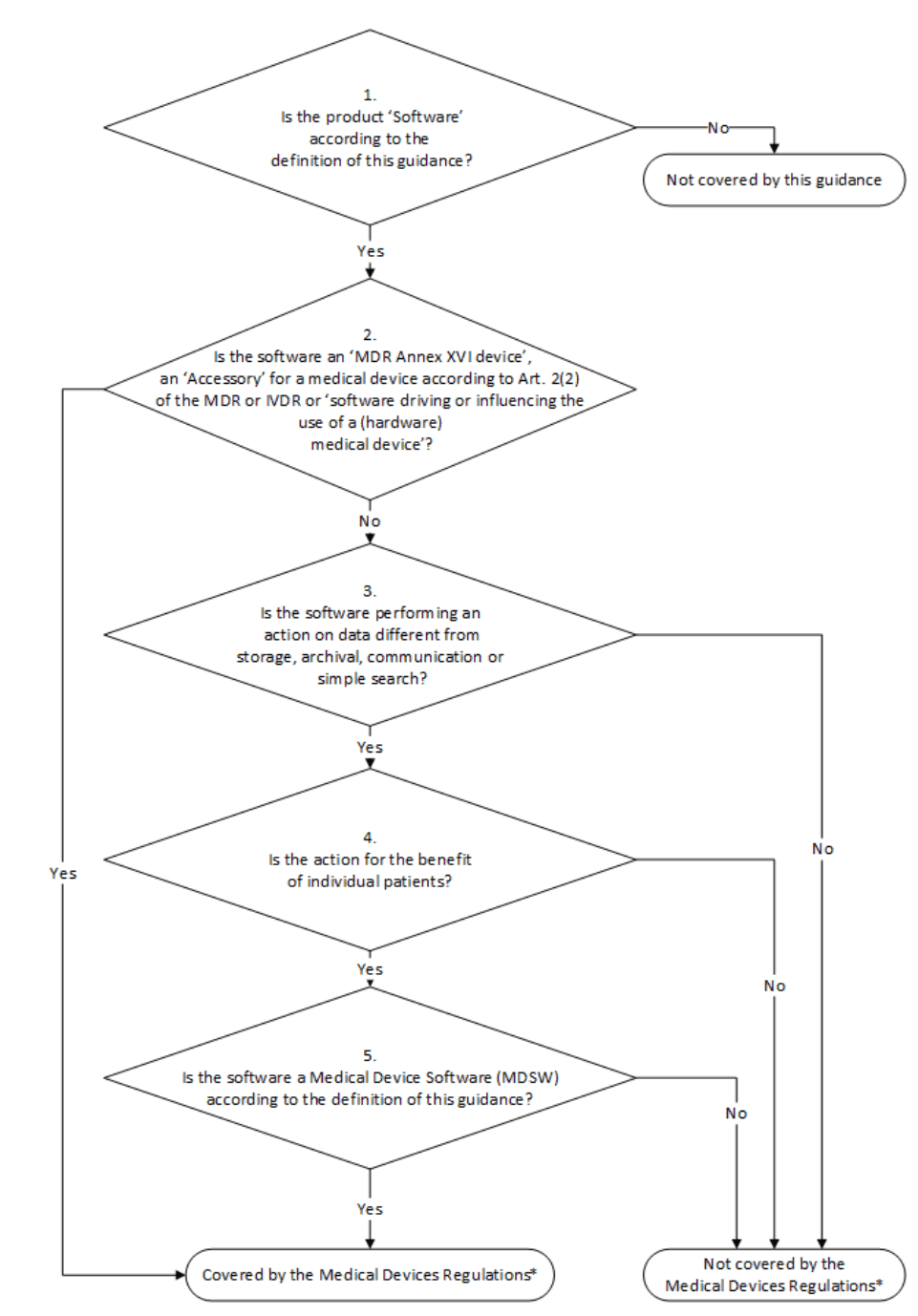
Besonders relevant ist die letzte Fallunterscheidung „Is the Software a Medical Device Software according to the definition of this Guidance?“
Zur Erinnerung:
„Medical device software is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a „medical device“ in the MDR or IVDR, regardless of whether the software is independent or driving or influencing the use of a device.“
MDCG 2019-11
Die MDCG weist darauf hin, dass es für die Klassifizierung als MDSW irrelevant ist,
- wo die Software läuft (in der Cloud, auf dem Smartphone, auf dem Server),
- wer die Software nutzt (Laien, Healthcare Professionals),
- ob die Software ein Medizinprodukt ansteuert oder nicht und
- ob die Software eigenständig läuft oder Teil eines Medizinprodukts ist.
b) Beispiele
Die MDCG nennt Beispiele für solche Medical Device Software (MDSW):
- Standalone-Software, die der Diagnose oder Behandlung dient, einschließlich Software zur Bewertung des Risikos einer Trisomie 21 eines Kindes basierend auf den Serummarkern der Mutter
- Software, die aus Ultraschallbildern und Laborparametern eines Patienten das Risiko von Prostatakrebs berechnet
- Software eines Massenspektrometers, die aus den Daten Mikroorganismen und deren Antibiotika-Resistenz feststellt
- App, die Alarme an den Patienten oder Arzt schickt, wenn sie aus unregelmäßigen Herzschlägen eine Arrythmie erkennt
- Software, die Glukosewerte misst, anhand derer die Insulindosis bestimmt und damit eine Insulinpumpe ansteuert (ggf. sogar als Closed-Loop-System)
- Cloud-basierte Software, die einen Point-of-Care-Test durchführt
- Bereitstellung von Expertenhilfe für klinische Entscheidungen (z. B. Planung der Strahlentherapie-Dosis)
Die MDCG nennt auch Beispiele für „Nicht-MDSW“:
- Sammeln und Verwalten von administrativen Patientendaten
- Speichern der Krankengeschichte von Patienten
- Ausführen von Rechnungs- und anderen Abrechnungsfunktionen
- Verbindung mit dem Sozialversicherungssystem für die Kostenerstattung
- Verbindung mit Systemen für die Verschreibung und Abgabe von Arzneimitteln
Lesen Sie auch unseren Beitrag, wann Software als IVD zählt. Diese Qualifizierung und Klassifizierung folgt ebenfalls aus dem MDCG-Dokument. Relevant ist auch die Einschätzung, wann Software in die Klasse I gemäß MDR fällt.
c) Sonderfall: Software nur zur Steuerung von Hardware
Wenn eine Software ausschließlich zur Steuerung der Hardware eines Medizinprodukts dient und keine eigenen medizinischen Zwecke verfolgt, ist sie nur als Zubehör dieses Medizinprodukts zu betrachten. Eine zusätzliche Klassifizierung gemäß Regel 11 kommt dann nicht in Betracht.
d) Kritik am MDCG-Dokument
Nur wenige Tage nach Veröffentlichung kam Kritik auf. Bei LinkedIn hat Professor Antonio Bartolozzi eine Präsentation publiziert:
Das Johner Institut schätzt diese Kritik wie folgt ein:
- Neue und nicht abgestimmte Definition
Es ist zutreffend, dass die neue Definition des Begriffs Medical Device Software (MDSW) von bestehenden Definitionen abweicht. Das kann Verwirrung stiften. Gleichzeitig stellt diese eigene Definition das eigentliche Konzept dar, das u.a. dazu führen kann und soll(!), dass Software, die Teil eines physischen Medizinprodukts ist, das ganze Medizinprodukt höher klassifiziert. - Neues Konzept
Weitere Kritikpunkte in der Präsentation scheinen dadurch bedingt zu sein, dass der Autor sich mit diesem Konzept nicht abfinden will oder es nicht versteht. Eine bewusste Abgrenzung und Gegenüberstellung der Konzepte hätte dem MDCG-Dokument gutgetan. - Verbindlichkeit der MDCG-Dokumente
Die Rechtsverbindlichkeit der MDCG-Dokumente zu hinterfragen, ist legitim. Hier werden unter Umgehung parlamentarischer Prozesse oder anderer Formen der Konsensbildung (wie bei der Normung) Regeln festgelegt, die de facto als verbindlich interpretiert werden. Sogar die EuGH-Richter haben sich auf solche Dokumente gestützt. - Beispiele
Weiter kritisiert Prof. Bartolozzi heftig die Beispiele. Natürlich werden Krankenhaus-Informationssysteme dazu genutzt, um Informationen für die Diagnose und Behandlung zu erhalten. Das steckt aber nicht in deren Zweckbestimmung. Dass diese Systeme folglich falsch klassifiziert sind, ist zutreffend. Das zu regeln, war aber nicht der Auftrag der MDCG.
Ebenso kann man zustimmen, dass ein PDMS ein Medizinprodukt ist. Hier windet sich die MDCG mit dem Hinweis auf Module etwas unglücklich aus der Sache. - Inkonsistenzen
In der Tat sollte die Klassifizierung von Medizinprodukten mit identischer Zweckbestimmung nicht davon abhängen, ob es Software- oder Hardware-Produkte sind.
Fazit: Die an manchen Stellen mit zahllosen Ausrufezeichen etwas unbeherrscht wirkende Kritik ist nicht ganz von der Hand zu weisen. Die Regel 11 ist in der jetzigen Form ungeeignet. Das MDCG-Dokument heilt diesen Missstand nur bedingt. Das liegt auch daran, dass es
- neue Definitionen und Konzepte einführt, ohne diese ausreichend zu erläutern und abzugrenzen,
- bestehende Konzepte (z. B. IMDRF) verbiegt,
- so formuliert, dass es nicht jeden Usability-Test bestehen würde, und
- an manchen Stellen zumindest etwas willkürlich bzw. als im Widerspruch mit vorhandenen Regeln stehend wirkt.
Die von Herrn Bartolozzi vorgeschlagene Klassifizierung hält das Johner Institut nicht für die Lösung. Demnach würden noch mehr Produkte in die Klasse III fallen, als dies die MDCG vorschlägt.
Er selbst argumentiert, dass die Klassifizierung risikobasiert erfolgen sollte, zieht sich dann aber auf die Regel 11 zurück, die nur die Schweregrade möglicher Schäden betrachtet.
Die Neuauflage des MDCG-Dokuments vom Juni 2025 ändert prinzipiell nichts an der Klassifizierung. Allerdings deutet die MDCG die „Prevention“ Richtung „Diagnose“ um:
a device intended to prevent the risk of illnesses or pathologies by […] can be considered as a device providing information which is used to take decisions with diagnosis purpose […] and in this case is in class IIa.
Die MDR weiß die Begriffe „Prevention“, „Diagnosis“, „Therapy“, „Prediction“ und „Prognosis“ sehr wohl zu unterscheiden.
Ein wenig mehr Klarheit geben die ergänzten Beispiele für Software als Medizinprodukt und Software, die nicht als Medizinprodukt gelten. Löblich zu erwähnen ist, dass das MDCG die Zweckbestimmung als Grundlage für Qualifizierung und Klassifizierung hervorhebt, auch wenn das eigene Ablaufdiagramm dem widerspricht. Dort wird anhand von Funktionalität („archive“, „storage“, „query“) die Entscheidung über die Qualifizierung empfohlen.
13. Australien
Im Dokument Exemption for Certain Clinical Decision Support Software Guidance on the exemption criteria beschreibt die australische TGA, unter welchen Umständen ein Clinical Decision Support System nicht der Regulierung unterliegt.
Das ist der Fall, wenn drei Bedingungen erfüllt sind:
- Die Software dient ausschließlich dazu, „Health Professionals“ Empfehlungen zur Diagnose, Verhütung, Heilung und Linderung von Krankheiten und Verletzungen zu geben.
- Sie analysiert oder verarbeitet nicht direkt medizinische Bilddaten oder Biosignale anderer Medizinprodukte (einschließlich IVD).
- Sie soll die Beurteilung durch Health Professionals bezüglich Diagnose oder Therapie nicht ersetzen.
Es gibt sehr viele Produkte, die von diesen Erleichterungen profitieren.
D) Beispiele für Software als Medizinprodukt
1. Webseite als Medizinprodukt
Fragestellung
Heise Online berichtete über ein Start-up, dessen Geschäftsidee darin bestand, Diagnosen von Hautkrankheiten zu erstellen. Die Webseite der Firma führte durch einen Fragebogen und erlaubte es, Fotos der eigenen Haut hochzuladen. Innerhalb von 48 Stunden erhielt man eine Antwort − das alles für nur 29 EUR. Die Webseite ist inzwischen offline.
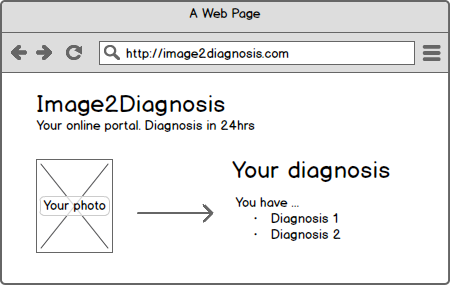
Es stellt sich schnell die Frage, ob eine/diese Webseite ein Medizinprodukt ist. Webseiten können generell Medizinprodukte sein. Und wie sieht es mit der konkreten Webseite aus? Fällt sie ebenfalls unter die Definition des Begriffs Medizinprodukt?
Bewertung
Die 2007/47 hat klargestellt, dass auch Standalone-Software unter die Definition Medizinprodukt fällt, wenn sie der Diagnose, Therapie und Überwachung von Krankheiten und Verletzungen dienen soll, um es etwas verkürzt wiederzugeben. Der Gedanke liegt nahe, dass die besprochene Webseite der Diagnose von Krankheiten dient, denn das Angebot ist die Diagnose von Hautkrankheiten.
Für die Klassifizierung (nicht nur von Webseiten) als Medizinprodukt ist die Frage, ob etwas passieren kann, nicht entscheidend.
2. Webservice als Medizinprodukt
a) Beispiel
Die Firma Promedas bot einen Webdienst an, der eine API für medizinische Diagnosen bietet.
Auf der Webseite des Herstellers hieß es:
„Promedas provides a clinical expertise system to medical professionals. The Promedas API can be integrated into existing medical systems that contain a patient file database. Using this data, Promedus can provide a differential diagnosis based on the data contained in a patient’s file. The API is currently in a developer beta. To access the API, contact Promedas.“
b) Fragestellungen
Damit ergeben sich gleich Fragen wie die folgenden:
- Ist dieser Webservice selbst schon ein Medizinprodukt?
- Wenn man diesen Service in eigene Software integriert, wie muss man ihn dann normgerecht behandeln? Als SOUP?
- Wenn dieser Webservice eine einfache GUI hat, kann er theoretisch weltweit aufgerufen und genutzt werden. Muss er dann automatisch für alle internationalen gesetzlichen Anforderungen ausgelegt werden? Auch wenn es einen Intended Use gäbe, dass der Dienst nur in der EU angewendet werden darf, dann würde im Rahmen der Marktbeobachtung sofort ersichtlich, dass er auch in den USA aufgerufen wird. Müsste er damit auch die FDA-Anforderungen erfüllen?
c) Bewertung
Frage 1: Bis vor kurzem hätte die Antwort lauten müssen „nein“. Der Webservice ist kein Medizinprodukt, weil er selbst nicht der Diagnose oder Behandlung von Patienten dient, sondern zur Integration in ein System vorgesehen ist, dessen Zweckbestimmung in der Diagnose oder Behandlung liegt.
Nach dem EuGH-Urteil (siehe unten) muss diese Einschätzung widerrufen werden. Demnach dürfen „Module“ eines Produkts als Medizinprodukt klassifiziert werden, auch wenn das Produkt, in das die Komponenten integriert sind, kein Medizinprodukt ist.
Es gibt noch weitere Möglichkeiten, um eine Komponente, die als Medizinprodukt klassifiziert ist, in ein Produkt zu integrieren, ohne dieses Produkt „zu infizieren“, d.h. dieses ebenfalls zum Medizinprodukt zu machen.
Wenn Sie weitere Informationen wünschen, dann nehmen Sie gerne Kontakt mit uns auf.
Frage 2: Es ist denkbar, einen Webservice als SOUP zu deklarieren und als Komponente, deren Entwicklung nicht den Anforderungen der IEC 62304 genügt, in ein Medizinprodukt zu integrieren. Im Rahmen des Risikomanagements wären dieses Vorgehen und diese Komponente natürlich zu bewerten.
Frage 3: Über einen Ausschluss in der Zweckbestimmung wäre man bereits aus dem Schneider. Wenn eine Behörde Maßnahmen androht, weil Kliniken oder Ärzte offensichtlich diese Vorgabe ignorieren, könnte man das über eine IP-Filterung einigermaßen gut lösen. Eine Behörde in Europa hat aber keine Handhabe in den USA und umgekehrt. Daher würde sie nie in einem anderen Rechtsbereich aktiv werden.
3. Dokumentenmanagementsysteme
a) Fragestellung
Im Rahmen des kostenlosen Micro-Consultings erreichte uns die Frage, ob Dokumentenmanagementsysteme (DMS) Medizinprodukte seien. Man argumentierte, dass durch Fehler in einem DMS Patienten zu Schaden kommen könnten. Andererseits würden aber immer Ärzte die letzte Entscheidung treffen.
b) Bewertung
Beide Überlegungen sind wichtig, aber für die Klassifizierung als Medizinprodukt nicht relevant:
Eine Software fügt nie direkt Schaden zu. Immer sind es Menschen oder Hardware, die letztlich den Patienten verletzen oder schädigen.
Relevant ist hingegen die Aussage des Herstellers, wofür seine Kunden das Produkt nutzen können und sollen. Wenn der Hersteller sagt, dass das System nur der Dokumentation dient, ist es kein Medizinprodukt. Wenn der Hersteller hingegen sagt, dass damit z.B. Röntgenbilder für die Verlaufskontrolle gespeichert werden sollen, dient das System der Therapie und ist damit definitionsgemäß ein Medizinprodukt.
Über die Formulierung dieser Zweckbestimmung kann der Hersteller sein Produkt zum Medizinprodukt machen oder eben nicht. Dabei ist die Zweckbestimmung nicht nur ein Dokument. Vielmehr dokumentiert der Hersteller dies auf die vielfältigste Weise:
- In der Gebrauchsanweisung
- In der Online-Hilfe des Produkts
- Auf seiner Webseite
- In Marketingmaterialien wie Flyern
- Auf Messen
- Sogar in Gesprächen
Daher empfiehlt das Johner Institut den DMS-Herstellern, sich eindeutig zu artikulieren. Das kann auch explizite Ausschlüsse von Funktionalitäten oder Anwendungsszenarien beinhalten.
Haben Sie Fragen zur Klassifizierung Ihres Produkts? Dann nutzen Sie unser kostenloses Micro-Consulting.
4. Krankenhaus-Informationssystem (KIS) als Medizinprodukt
a) Fragestellung
Die Aufgabe klinischer Informationssysteme (KIS) besteht offensichtlich darin, die Anwender bei der Diagnose, Überwachung und Behandlung von Patienten zu unterstützen. Dass diese Systeme auch gelegentlich Patienten (indirekt) gefährden, ist ebenfalls nicht unbekannt. Das kann der Fall sein, wenn sie
- Patientendaten vertauschen,
- Daten verlieren,
- sehr langsam auf Benutzereingaben reagieren (z.B. bei einem Malware-Befall),
- überhaupt nicht mehr reagieren, nicht mehr starten, komplett ausfallen oder
- Informationen nicht an andere Systeme weiterleiten.
b) Bewertung
Entscheidend für die Frage, ob diese Software als Medizinprodukt zu klassifizieren ist, ist auch hier nicht die die Gefährdung, sondern nur die Übereinstimmung der Zweckbestimmung mit der Definition des Begriffs Medizinprodukt.
- Ein System, das nur zur Dokumentation oder Abrechnung vorgesehen ist, fällt nicht darunter.
- Ein System, das aus eingegebenen Werten Diagnose- oder Therapieempfehlungen ableitet, das Warnungen beispielsweise bei Medikamentenwechselwirkungen gibt, schon.
c) Konsequenzen für die Betreiber
Da auf Dauer kein KIS-Hersteller überleben wird, dessen Systeme nichts anderes können, als eingegebene Daten wieder anzuzeigen, werden mittelfristig alle KIS zum Medizinprodukt. Das will nur keiner hören, besonders nicht die Betreiber, nämlich die Krankenhäuser.
Der Grund ist offensichtlich: Sobald ein KIS als Medizinprodukt klassifiziert ist, unterliegt es der Medizinprodukte-Betreiberverordnung. Und diese verlangt u. a.:
- Personen, welche das System anwenden, betreiben und instand halten, müssen geschult sein. Das gilt es zu dokumentieren.
- Das System muss regelmäßig überprüft werden, spätestens alle zwei Jahre.
- Nach jeder Instandhaltungsmaßnahme (Software-Update) müssen alle sicherheitsrelevanten Funktionen und Konstruktionsmerkmale geprüft werden.
Doch wie sollen Krankenhäuser dies bewerkstelligen? Wie sollen sie vollständig prüfen, dass nach einem Software-Update ein KIS mit Diagnosefunktionalität noch funktioniert? Dass es mit dem RIS, das im gleichen Netzwerksegment hängt, noch fehlerfrei zusammenarbeitet? Dass es keine Rückwirkungen auf Ultraschallgeräte gibt, die ebenfalls ans Netzwerk angeschlossen sind?
Die einzige Möglichkeit, diese nahezu unendlichen Kombinationen möglicher Fehlerursachen anzugehen, besteht darin, ein Risikomanagement zu betreiben, mit dem sich die Prüfschritte priorisieren lassen.
Einige Firmen klassifizieren ihre Systeme als Medizinprodukt, wie z. B. GE Healthcare sein Informationssystem Centricity, das die CE-Kennzeichnung (gemäß MDD, 93/42/EC) erhielt. Hierzu die entsprechende Pressemeldung von GE.
5. Software als IVD
Software kann nicht nur als Medizinprodukt (gemäß MDD/MDR), sondern auch als IVD (gemäß IVDD/IVDR) klassifiziert werden.
Lesen Sie hier mehr zum Thema IVD Software.
6. ChatGPT als Medizinprodukt
Dass ChatGPT auch auf medizinische Fragen Antworten gibt, wissen oder ahnen Sie. Eine Kanzlei kommt deshalb zu der Schlussfolgerung:
„Unserer Rechtsauffassung nach fällt diese Software in Deutschland und in Europa unter die Regulation als Medizinprodukt“
Apotheke Adhoc
Auch die E-HEALTH-COM berichtet, dass eine „auf digitale Medizinprodukte spezialisierte Kanzlei“ beim BfArM „die regulatorische Einordnung der KI-basierten Software ChatGPT als Medizinprodukt gefordert“ hat.
Als Grund wird die Erkenntnis genannt, dass „ChatGPT aufgrund seiner diagnostischen und therapeutischen Nutzungsmöglichkeiten in den Anwendungsbereich der Medical Device Regulation (MDR) fällt“.
Das Johner Institut stimmt zu, dass ChatGPT diagnostisch und therapeutisch genutzt werden kann. Hingegen kann es die Schlussfolgerung nicht nachvollziehen, dass ChatGPT bereits durch die Möglichkeit, so genutzt zu werden, zum Medizinprodukt wird.
Entscheidend ist die Zweckbestimmung des Herstellers. Es ist davon auszugehen, dass OpenAI seine Produkte nicht dazu vorsieht, diagnostisch oder therapeutisch genutzt zu werden. In seinen Terms of use schreibt die Firma:
„You must not use any Output relating to a person for any purpose that could have a legal or material impact on that person, such as […] medical, or other important decisions about them.”
Wenn hingegen ein Medizinprodukt ChatGPT nutzt, dann muss das ganze Produkt die regulatorischen Anforderungen erfüllen. Zwar ließe sich ChatGPT als SOUP deklarieren; aber Spezifikation und Verifikation dieser Komponente dürften ebenso schwierig werden wie der Nachweis eines positiven Nutzen-Risiko-Verhältnisses.
Man darf gespannt sein, ob und wie das BfArM reagiert. Die Durchsetzung der gesetzlichen Anforderungen liegt bei den Ländern …

Zertifizieren Sie Ihre Software mit Leichtigkeit als Medizinprodukt
Entdecken Sie jetzt, wie Sie Ihre Software als Medizinprodukt zertifizieren lassen können. Erfahren Sie alles über die IEC 62304 Norm und lernen Sie, wie Sie die Anforderungen einfach und praxisorientiert umsetzen können. Mit über 25 Videos und einem kompletten Set einer IEC 62304-konformen Software-Akte unterstützen wir Sie dabei, Ihre Software erfolgreich zu zertifizieren.
7. Weitere Beispiele
Viele weitere Beispiele enthält der Anhang I des Guidance-Dokuments der MDCG:
- Hospital Information Systems
- Decision Support Software
- Electronic Patient Record Systems
- Clinical Information Systems – CIS/Patient Data Management Systems – PDMS
- Pre-hospital Electrocardiograph (ECG) System
- Picture Archive Communication System (PACS)
- Telemedicine systems
- Telesurgery
- Web systems for monitoring of data
- Laboratory Information Systems (LIS) and Work Area Managers (WAM)
- IVD: Expert system
- IVD: Interpretation of raw data
- IVD: Home care monitoring, wired or mobile
- IVD: Image Management System (IMS)
E) Sonderfall: Komponenten als Medizinprodukt
1. MEDDEV 2.1/6: Nur „medizinische Module“ müssen MDD erfüllen
Die EU-Kommission hat im Juli 2016 die Leitlinie MEDDEV 2.1/6 (siehe unten) mit dem länglichen Titel „Guidelines on the Qualification and Classification of Stand Alone Software used in the Healthcare within the Regulatory Framework of Medical Devices“ in einer zweiten Version veröffentlicht.
Dieses Dokument stellt fest, dass eine Software aus Modulen bestehen kann. Nur die Module müssten den Anforderungen der Medizinprodukterichtlinie genügen, die eine entsprechende medizinische Zweckbestimmung verfolgen. So steht geschrieben:
„Some stand alone software may break down into a significant number of applications for the user where each of these applications is correlated with a module. Some of these modules have a medical purpose, some not. The modules which are subject to the medical device Directives (Figures 1 and 2) must comply with the requirements of the medical device Directives and must carry the CE marking. The non-medical device modules are not subject to the requirements for medical devices.“
Die MDCG verfolgt diesen Ansatz weiter. Sie schreibt:
„Computer programmes used in healthcare can have applications which consist of both medical device and non-medical device modules.
MDCG 2019-11
The modules which are subject to the Medical Devices Regulations must comply with the requirements of the medical device regulations and must carry the CE marking. The non-medical device modules are not subject to the requirements for medical devices. It is the obligation of the manufacturer to identify the boundaries and the interfaces of the different modules.“
Ein Beispiel für solch eine Software könnte ein Krankenhaus-Informationssystem darstellen, das aus Nichtmedizinprodukte-Modulen (Abrechnung, Stammdatenverwaltung usw.) und Medizinprodukte-Modulen (z. B. Arzneimittelprüfung, radiologische Befundung) besteht.
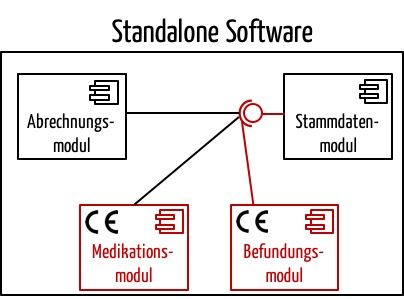
Lesen Sie hier mehr zum Thema Software-Module bzw. Software-Komponenten.
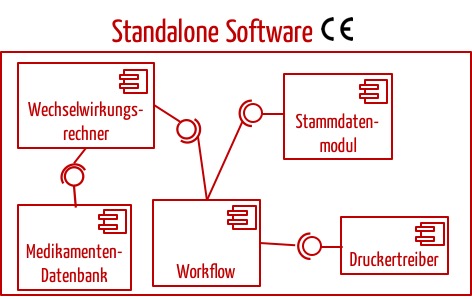
In den meisten Fällen wird man die Standalone-Software als Ganzes als Medizinprodukt zertifizieren und nicht deren einzelne Komponenten. Beispielsweise würde man bei einer Software zur Arzneimittelsicherheit nicht anstreben, einzelne Module zu zertifizieren.
2. Bestätigung durch das EuGH
Ein EuGH-Urteil vom Dezember 2017 bestätigt die Möglichkeit, Software-Module mit/ohne Medizinprodukteigenschaft abzugrenzen. Der Europäische Gerichtshof schreibt im Absatz 36 klar:
„Im Fall einer medizinischen Software, die gleichzeitig Module umfasst, die der Definition von ‚Medizinprodukt‘ entsprechen, und andere, die ihr nicht entsprechen und kein Zubehör im Sinne von Art. 1 Abs. 2 Buchst. b der Richtlinie 93/42 sind, fallen nur die erstgenannten Module in den Anwendungsbereich dieser Richtlinie und müssen mit einer CE‑Kennzeichnung versehen werden.“
Die Richter interpretieren die Leitlinie MEDDEV 2.1/6 sogar wie folgt:
„In diesen Leitlinien heißt es weiter, dass Software, die auf die Daten in keiner Weise einwirkt oder die sich auf die Speicherung, Archivierung, verlustfreie Komprimierung oder letztlich auf die einfache Suche beschränkt, d. h. in diesem letzteren Fall Software, die eine Datenbankfunktion hat und anhand derer aus Metadaten stammende Informationen gefunden werden können, ohne sie zu verändern oder sie zu interpretieren, nicht als Medizinprodukt angesehen werden darf.“ (Hervorhebung durch Johner Institut)
Weiterhin steht in dem Urteil:
„Insoweit bestätigen die in Rn. 33 des vorliegenden Urteils genannten Leitlinien der Kommission in Titel 4 („Module“) im Wesentlichen, dass, wenn eine Software aus Modulen besteht, die der Definition des Begriffs „Medizinprodukt“ entsprechen, und solchen, die dieser Definition nicht entsprechen, nur die erstgenannten Module mit einer CE‑Kennzeichnung versehen werden müssen, da die anderen den Bestimmungen dieser Richtlinie nicht unterliegen. Diese Leitlinien stellen klar, dass es Sache des Herstellers ist, die Grenzen und Schnittstellen der verschiedenen Module anzugeben. Diese müssen, was die der Richtlinie 93/42 unterliegenden Module anbelangt, vom Hersteller eindeutig angegeben werden und auf der künftigen Verwendung des Produkts beruhen.
Der Hersteller einer solchen Software ist daher verpflichtet, diejenigen Module anzugeben, die Medizinprodukte darstellen, damit die CE‑Kennzeichnung allein auf diesen Modulen angebracht werden kann.“
Sie können sich das Urteil hier herunterladen.
Zusammenfassung
- Eine Software kann aus verschiedenen Modulen bestehen, bei denen nur bestimmte Module als Medizinprodukte zu zertifizieren sind.
- Der Hersteller muss diese Module angeben und auf diesen Modulen eine CE-Kennzeichnung anbringen.
- Der Hersteller muss die Grenzen zwischen diesen Modulen klar identifizieren, wobei die Aufteilung auf der Zweckbestimmung zu basieren hat (s. Seite 18, vorletzter Absatz der MDDEV 2.1/6).
Das Urteil sollte nicht so verstanden werden, dass Standalone-Software immer in Module zerlegt werden und dann pro Modul entschieden werden muss, ob es ein Medizinprodukt ist. Die Zerlegung der Software in Komponenten fordert zwar die IEC 62304 (außer für Klasse-A-Software). Aber Hersteller sollten sehr sorgfältig entscheiden, ob sie tatsächlich einzelne Module als Medizinprodukte zertifizieren lassen. In den meisten Fällen rät das Johner Institut davon ab (s.u. und Beispiel in Abb. 3).
3. Bewertung
a) Rechtssicherheit liegt vor
Das Johner Institut vertrat wie einige Benannte Stellen die Meinung, dass Hersteller ein Produkt entweder als Medizinprodukt oder als Nicht-Medizinprodukt in Verkehr bringen können. Es vermutete, dass die Autoren der MEDDEV 2.1/6 die Begriffe wie „Modul“ anders verstehen als Software-Entwickler. Sätze wie der folgende verstärkten den Eindruck einer eigenen Begriffswelt:
„Computer programmes(sic!) used in healthcare mostly have applications which consist of both medical device and non-medical device modules.“
Die EuGH-Richter sind jedoch der MEDDEV gefolgt, sodass nun eine klare Rechtssprechung vorliegt. Die Definition dessen, was ein Modul ist, haben die Richter aber nicht geliefert. Sind es Komponenten, wie sie ein Software-Entwickler versteht, die (unverzichtbarer) Teil einer Software sind? Oder sind es Plugins, die man zusätzlich und optional installieren kann?
Man muss aber auch den Hintergrund des Urteils verstehen: Der französische Staat will eine bestimmte Software regulieren. Philips möchte ihm das nicht zugestehen, weil das Produkt CE-gekennzeichnet und somit von weiterer Regulierung ausgeschlossen ist. Der Gerichtshof sagt: In diesem Fall hat der französische Staat recht, weil die Software zum großen Teil kein Medizinprodukt ist. Also ganz speziell für ein Produkt, ein KIS, bei dem wirklich große Teile, die auch separierbar sind, kein Medizinprodukt sind.
b) Vorteile durch das Urteil
Herstellern von Standalone-Software können mit Veröffentlichung des Urteils von der Möglichkeit Gebrauch machen, nur die Module einer Software als Medizinprodukt zu zertifizieren, die über eine entsprechende Zweckbestimmung verfügen. Das empfiehlt sich in folgenden Situationen:
- Es gibt im Vergleich zur gesamten Software nur wenige oder/und nur kleine Module mit einer medizinischen Zweckbestimmung.
- Die Module sind nach (medizinischen) Funktionen gebildet und ausreichend stark voneinander abgetrennt.
- Der Aufwand für die Zertifizierung der gesamten Software ist für den Hersteller nicht leistbar.
Das Johner Institut erkennt einige Gründe an, die für die Entscheidung des EuGH sprechen:
- Die Entscheidung kann im Hinblick auf eine Deregulierung und als Gegengewicht zur fatalen Regel 11 der MDR für Hersteller hilfreich sein.
- Sie folgt dem Trend, dass sich Medizinprodukte zunehmend öffnen bis „auflösen“ und Betreiber die Komponenten (verschiedener Hersteller) zu Systemen kombinieren.
- Die Hersteller werden damit stärker gezwungen, in funktionalen und schwach gekoppelten Komponenten zu denken. Das verbessert die Software-Architektur und damit die Stabilität, Wartbarkeit und Testbarkeit.
c) Herausforderungen und Risiken
Hersteller, die diese Möglichkeit nutzen, sollten sich jedoch der Konsequenzen bewusst sein:
- Risikomanagement
Es dürfte zumindest schwierig werden, im Risikomanagement zu argumentieren, dass die Risiken beherrscht sind, wenn die Daten, die ein zertifiziertes Modul nutzt, von Modulen stammen, über deren Güte keine ausreichende Aussage getroffen werden kann, weil die Dokumentation des jeweiligen Software-Lebenszyklus (gemäß IEC 62304) nicht vorliegt. - Usability
Die Gebrauchstauglichkeit eines Moduls lässt sich in der Regel nicht einzeln bewerten. Ob die Prüfung eines „Fensters“, eines „Screens“ oder einer Maske (ohne den Kontext der gesamten Software zu betrachten) verlässliche Ergebnisse liefert, erscheint fraglich. Die IEC 62366-1 verlangt, die Gebrauchstauglichkeit entlang der Benutzungsszenarien zu bewerten und nicht für einzelne technische Module oder UI-Elemente. - „Audit-Sicherheit“
Software-Entwickler werden anfälliger für Fragen von Auditoren, ob der gerade entwickelte Code zu einem zertifizierten oder zu einem nicht zertifizierten Modul gehört. Viele Entwicklungsabteilungen empfinden bereits Stolz, wenn es gelingt, einen einzigen Satz an „Best Practices“ zu verankern … - Regulatorische Aufwände
Wenn mehrere Module, die ein Hersteller gemeinsam in Verkehr bringt, Medizinprodukte sind: Liegt dann gar ein System oder eine Behandlungseinheit vor? Bedarf es einer entsprechenden Erklärung? Dann müsste der Hersteller nicht ein Produkt registrieren, sondern n Module und zusätzlich ein System bzw. eine Behandlungseinheit. - Software-Lebenszyklus und Dokumentation
Die Aufteilung in Module, die jeweils einzelne Medizinprodukte sind, ermöglicht es, die Module auch unabhängig voneinander zu entwickeln. Doch dies erfordert für jedes Medizinprodukt eine eigene Dokumentation sowie eine eigene Zweckbestimmung, Risikomanagementakte, Software-Lebenzyklusakte, Konformitätserklärung, Handbücher/Gebrauchsanweisungen, klinische Bewertung usw. Über die Herausforderungen beim Usability Engineering wurde bereits gesprochen. - Labeling
Wo man die CE-Kennzeichnung im Fall von einzelnen Modulen konform mit den Anforderungen der MDD und MDR anbringen kann und soll, haben die Richter nicht diskutiert. Wie soll das bei einem Modul ohne Benutzerschnittstelle erfolgen?
Beachten Sie: Wenn die Hersteller diese Dokumente nicht für jedes Modul und damit n-fach erstellen möchten, wenn sie nicht n-mal eine Post-Market Surveillance durchführen und dokumentieren wollen, müssen sie die Software weiter als ein einziges Produkt in den Verkehr bringen. Doch was haben sie dann gespart? Sie haben eine komplette technische Dokumentation erstellt, die es bei der Zulassung eines Medizinprodukts bedarf.
Die Möglichkeit, die Software-Dokumentation auf die „kritischen“ Module/Komponenten zu beschränken, gibt die IEC 62304 bereits heute. Dass eine möglicherweise einfachere Argumentation bezüglich Segregation der Software-Komponenten die zusätzlichen Aufwände für die Inverkehrbringung von einzelnen Modulen als Medizinprodukt rechtfertigt, kann bezweifelt werden.
Offen bleibt noch die Frage, ob auch das die Komponenten umgebende Produkt ein CE-Zeichen bekommt. Die MDCG schreibt:
„This raises the issue as to whether the whole product can be CE marked when not all applications have a medical purpose.“
MDCG 2019-11
d) Eine Verschiebung der Ebenen
Wer es absurd findet, dass ein Software-Modul ein Medizinprodukt sein kann, möge sich an die ersten Diskussionen erinnern, ob eine Standalone-Software ein Medizinprodukt sein kann. Denn eine Standalone-Software kann ohne eine Plattform (Hardware, Betriebssystem, ggf. ‚Runtime Environment‘) nicht funktionieren und benötigt die technischen Interfaces der Plattform (Monitor, Tastatur, Touch, Datenschnittstellen …). Niemand stellt inzwischen infrage, dass eine Standalone-Software ein Medizinprodukt sein kann.
Für ein Software-Modul als Medizinprodukt wird die Grenze zwischen Medizinprodukt und Umgebung (Plattform) um eine weitere Ebene verschoben.
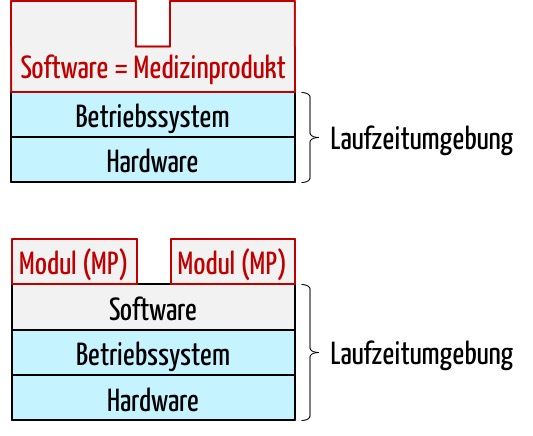
Das Modul als Medizinprodukt benötigt als erste Umgebungsebene nun das proprietäre Softwaresystem, das wiederum das Betriebssystem und dieses wiederum die Hardware. Die technische Dokumentation für die Module (= Medizinprodukte) muss diese erweiterte Laufzeitumgebung spezifizieren.
Bei der Verifizierung, dem Testen, muss in Analogie zur Standalone-Software auch das Softwaresystem berücksichtigt werden, so wie man das Betriebssystem und die Plattform beim Standalone-Softwareprodukt berücksichtigen muss.
e) Fazit
Die FDA hat längst mit der Deregulierung begonnen. Dass aus Europa (endlich) ein vergleichbares Signal kommt, ist gut. Die Entscheidung des EuGH eröffnet Herstellern neue Möglichkeiten und erleichtert es, Produkte und Systeme aus Modulen zu kombinieren.
Ob der Aufwand für die Zulassung von einem oder mehreren Modulen als Medizinprodukt tatsächlich geringer ist als der Aufwand für die Zulassung der gesamten Software (die diese Module enthält), ist fraglich und im Einzelfall zu prüfen. In der Regel sei von dem Weg, den der EuGH eröffnet hat, abgeraten.
Es ist zu befürchten, dass die Motivation des Urteils zu einer Entscheidung geführt hat, die in der Praxis mehr Probleme aufwerfen wird, als dass sie Erleichterung verschafft.
Änderungshistorie
- 2026-01-13: Abschnitt C) 8.) („Swissmedic“) aktualisiert: Es gilt ein neues Merkblatt.
- 2025-07-09: Links zu neuer Version der Leitlinie MDCG 2019-11 gesetzt, Abschnitt C) 5. (MEDDEV) und C) 12. (MDCG) überarbeitet
- 2025-02-19: Absatz in C.2 sowie mehrere Links auf offizielle Webseiten ergänzt
- 2023-11-17: Absatz mit ChatGPT aktualisiert aufgrund der Meldung von E-Health-Com
- 2023-02-03: Absatz mit ChatGPT eingefügt. Redaktionelle Änderungen
- 2022-16: Unter „Entscheidungshilfen“ Digital Health Policy Navigator verlinkt
- 2022-09: Unter C) aktuelles MHRA-Dokument ergänzt
- 2022-09: Unter C) 13 Guidance der australischen TGA ergänzt
- 2022-08: Abschnitt „B) Qualifizierung / Klassifizierung von Software als Medizinprodukt“ aktualisiert
- 2020-12: Im Kapitel „13. Australien“ die Ergebnisse des Feedback-Prozesses eingearbeitet



Hallo Herr Johner,
vielen Dank für die Zusammenfassung der Dokumente. Leider ist der Link zu http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-131209-samd-key-definitions.pdf nicht. Über http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-131209-samd-key-definitions-140901.pdf#search=„samd key definitions“ habe ich es dann gefunden.
Viele grüße,
Pierre Baum
Hallo Herr Prof. Johner,
die Auseinandersetzung mit den unterschiedlichen Anleitungen, wie nun die Produkte zu klassifizieren sind, ist sehr spannend – vor allem unter dem Aspekt, wie das wohl eine Benannte Stelle sieht.
Laut EU Kommission sind die MEDDEV Guidelines rechtlich nicht bindend.
Das BfARM hat aktuell auf seiner Webseite https://www.bfarm.de/DE/Medizinprodukte/Abgrenzung/MedicalApps/_node.html zur Klassifizierung auf Anhang IX der Richtlinie 93/42/EWG verwiesen.
Wenn wir dementsprechend in diesem Jahr/Anfang 2017 ein neues Produkt als Klasse I einstufen und entsprechend in den Markt bringen, machen wir dann etwas falsch? Gibt es auch bei den Guidelines eine Übergangsfrist, auf die man sich berufen kann?
Vielen Dank und viele Grüße
Regina Preysing
Liebe Frau Preysing,
Stand Dezember 2017 gibt es nur den Anhang IX. Bitte ignorieren Sie noch alle künftigen Klassifizierungen der MDR. Noch ist die nicht verabschiedet. Das wird voraussichtlich Frühsommer 2017. Natürlich sollte man sich auf die MDR vorbereiten. Das wird mehr Arbeit als uns allen lieb ist. Aber noch keine Produkte anders klassifizieren.
Herzliche Grüße, Christian Johner
Hallo,
meiner Meinung nach steht in der „COICR Contribution“ tatsächlich nichts neues, das ist im Grunde nur die Meddev 2.1/6 noch mal erläutert/abgeschrieben ….
viele Grüße
Christian
Hallo Herr Prof. Johner,
ihre Beiträge samt scharfsinnigen Kommentaren sind sehr informativ und auf dem Punkt! Vielen dank!
Es gibt eine Kategorie von SW, die in keinem Dokument erwähnt wird, zumindest meiner Verständnis nach. Der Unterschied zwischen Embedded und Stand-Alone Software als MD ist mir klar.
Was ist aber mit einem SW-Tool, welches kein MD ist, und nicht von Kunden sondern nur von Service Technikern eingesetzt wird um die SW eines MD zu verändern. Konkretes Beispiel: Ein selbst entwickeltes SW-PC-Tool für die
a. Durchführung eines SW-Updates oder
b. Änderung von individuellen Parametern (Konfiguration-Kalibrierung).
Fällt es unter der Kategorie „Accessory“ obwohl es nicht an Kunden verkauft wird?
Vielen Dank im Voraus
Christos Freris
Sehr geehrter Herr Feris,
bei einem solchen Tool handelt es sich in der Tat um ein Zubehör. Allerdings liegt bei Ihnen keine Inverkehrbringung vor. Entsprechend muss kein Konformitätsbewertungsvefahren durchlaufen werden. Die grundlegenden Anforderungen müssen aber erfüllt sein.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
vielen Dank für die prompte Reaktion! Ich wollte Ihre Meinung wissen, welche absolut Sinn macht.
Leider war der FDA-Inspektor der Ansicht, es sei „part of the medical device, because it changes the configuration or the whole SW on the device“ und verpasste uns eine 483, weil das Tool als SW Class A von uns eingestuft war anstatt Class C (die der SW des Gerätes). Widerstand zwecklos!
Vielen Dank
Christos Freris
Das tut mir leid, lieber Herr Freris!
Die SW ist ein Zubehör, da irrt der Inspektor wahrscheinlich. Allerdings hat er mit der Klassifizierung möglicherweise Recht. Wenn die SW durch eine Fehlkonfiguration einen nennenswerten Schaden zur Folge haben kann, dann ist die SW Klasse C.
Dass der Inspektor überhaupt von Sicherheitsklassen nach IEC 62304 spricht, überrascht mich. Haben Sie Konformität mit diesem Standard erklärt? Das wäre nicht notwendig gewesen. Oder geht es um den Level of Concern?
Beste Grüße, Christian Johner
Vielen Dank für den sehr interessanten Artikel. Die Ausführungen zu den Modulen deckt sich meines Erachtens auch sehr gut mit der FDA (draft) Guidance vom April 2018: „Multiple Function Device Products: Policy and Considerations“
darin schließt die FDA auch die Bewertung von nicht medizinischen Modulen / Funktionen aus.
Quelle:
https://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM605683.pdf
Danke für diese Ergänzung, Herr Marzinko!
Eine kurze Beschreibung, was dieses Dokument aussagt finden Sie auch hier.
Newsletter Abo
Sehr geehrter Herr Prof. Johner,
vielen Dank für den wie immer sehr interessanten Artikel. Eine Frage: Wie muss man sich die CE-Kennzeichnung eines Software-Moduls vorstellen? Hier fehlt mir völlig die Vorstellung.
Für einen Hinweis wäre ich Ihnen sehr dankbar.
Herzliche Grüße
Roland Weghorn
Sehr geehrter Herr Weghorn,
das ist auch schwer vorstellbar. Genau diese Frage stellt der Artikel auch. Ggf. könnte es der Splash-Screen oder das Help>About der übergeordneten Anwendung einen Ansatz bieten.
Viele Grüße, Christian Johner
Vielen Dank Herr Prof. Johner. Wenn das reicht, ist alles gut.
Sehr geehrter Herr Prof. Johner,
wir nutzen in der Entwicklung unserer Medizinproduktesoftware Developer-Express-Komponenten. Würden Sie diese eher als zur Laufzeitumgebung (ähnlich wie das .net-Framework) gehörend oder eher als SOUP sehen?
Und wie verhält es sich mit Angular (Laufzeitumgebung)?
Viele Grüße,
Thomas Hertwig
Lieber Herr Hertwig,
die formale Antwort ist die: Wenn Sie die Komponenten als Teil Ihres Produkts mit ausliefern, sind das SOUP.
Die inhaltliche Antwort: Die DevExp-Komponenten und Angular sind fast immer Teil des Produkts, das setzt man nicht als bereits installiert voraus. Einmal sind diese Komponenten zu spezifisch, zum anderen wollen Sie sicher sein, dass genau die richtigen Versionen genutzt werden. Also: SOUP.
Die praktische Antwort: ein Auditor wird das in der Regeln nicht unterscheiden können.
Viele Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
vielen Dank für Ihren ausführlichen und äußerst interessanten Artikel zum Thema Software als Medizinprodukt.
Wir als IVD-Hersteller stehen im gegenwärtigen Entwicklungsprozess genau vor diesem Problem. Für einen unserer Analyzer gibt es künftig eine neue Software. Damit auch Geräte im Feld von der neuen Software profitieren, wird unsererseits ein Software-Update ermöglicht. Realisiert wird dieses Software-Update durch eine eigens geschriebenen Software, die keinen medizinischen Zweck hat, sondern lediglich eine Update-Funktion. Zudem überprüft sie vor dem Start des Updates, ob die Einstellungen des Analyzers (Datum, Parameter) korrekt sind. Wenn nicht, werden diese Parameter durch die Software entsprechend korrigiert. Somit würde sie in ein Medizinprodukt eingreifen.
Müssen wir die Update-Software als Medizinprodukt klassifizieren, oder genügt die Begründung, dass die Software keinen medizinischen Zweck erfüllt?
Ich danke Ihnen im Voraus für eine Einschätzung.
Herzliche Grüße
Anne-Kathrin Lerke
Sehr geehrte Frau Lerke,
danke für die spannende Frage! Wenn Sie eine Software schreiben, die speziell dafür gedacht ist, die Software auf Ihrem Medizinprodukt zu aktualisieren, ist das ein Zubehör. Für Zubehör gelten leider die gleichen Regularien wie für Medizinprodukte.
Viele Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
ich danke Ihnen für die schnelle Antwort, die das bestätigt, was unser RA-Kollege bereits vermutet hat. Insgeheim hatte natürlich noch gehofft, weder in die Kategorie Medizinprodukt noch in die Kategorie Zubehör zu fallen.
Viele Grüße
Anne-Kathrin Lerke
Guten Tag,
mobile Produkte, die sogen. wearables sind in aller Munde. Diese oft als smart watch ausgeführten, multifunktionalen Geräte können in der einfachsten Version manuell oder automatisch Notfall-Alarmierungen auslösen.
Ist nach der neuen MDD eine smart watch, die eine Alarmierung bei einem Notdienst auslöst, wenn ein Senior selbst den button drückt oder z.B. per Areal-Überwachung(GPS) einen Dementen außerhalb des Areals erfaßt, damit schon ein Medizinprodukt?
Anders gefragt – wird eine einfache chinesische tracker smart watch, welche nach LVD, EMV und RED die EU-Konformität hatte, durch Implementation einer Senioren-Überwachungs app durch eine Software-Firma in der EU zum Medizinprodukt? Die Konsequenzen einer Fehlfunktion des Geräts könnten sich durchaus lebensbedrohlich auswirken.
Oder bleibt die Konformität der „hardware“ inkl. Betriebssystem zur Kommunikation und Positionsüberwachung nach LVD, EMV und RED bestehen und es muß nur die zugefügte app einer Neubewertung nach MDD unterzogen werden?
MfG
Rainer Gutschmidt
CE-Koordinator
Sehr geehrter Herr Gutschmidt,
danke für Ihre Frage zu Klassifizierung und das anschauliche Beispiel!
Die Klassifizierung hängt nicht primär von der Funktionalität, sondern von der Zweckbestimmung ab. Wenn die Zweckbestimmung darin besteht, dass ein dementer Patient überwacht werden soll, dann liegt die Zweckbestimmung in der Überwachung einer Krankheit. Genau das würde unter die Begriffsdefinition „Medizinprodukt“ fallen.
Eine hierfür relevante Änderung der Definition zwischen MDD und MDR (die meinten Sie, oder?), gibt es nicht.
In dem genannten Fall wäre die App das Medizinprodukt, weil die App und nicht die Hardware für die o.g. Zweckbestimmung in den Verkehr gebrach wird. Allerdings muss der Hersteller die Risiken, die sich aus diese Hardware ergeben berücksichtigen und beherrschen.
Der Hersteller ist nicht verpflichtet, für die Hardware (SmartPhone) die EMV zu gewährleisten, da er das Produkt nicht in den Verkehr bringt.
Viele Grüße, Christian Johner.
Guten Tag,
Bei der Klassifikation von Produkten als Medizinprodukt ja / nein (MEDDEV 2.1/6 spricht hier von Qualification, passt für mich besser als der Begriff Klassifikation, aber leider ist die MEDDEV 2.1/6 nicht mehr anwendbar mit der MDR) sehe ich bei Medizinproduktherstellern immer wieder, dass das Risiko, ob ein Patient geschädigt werden kann, einfliesst in die Entscheidung, ob es sich um ein Medizinprodukt handelt oder nicht. Ich kenne keine einzige Stelle in den einschlägigen Guidelines und Regularien, wo das Risiko als Faktor für die Qualifikation herangezogen werden kann.
Vielleicht habe ich das eine wichtige Dokument nicht gelesen oder den Inhalt nicht verstanden. Können Sie mir einen Hinweis dazu geben, ob das Risiko an sich ein Faktor bei der Qualifikation sein kann oder nicht?
Besten Dank und freundliche Grüsse
Peter Egli
Sehr geehrter Herr Egli,
danke für die spannende Frage! Sie haben Recht, dass die MEDDEVs unter der MDR nicht mehr gültig sein werden. Ich erwarte allerdings, dass man sich weiter an diesen Leitlinien orientiert, bis man etwas Neues hat.
Dass das Risiko alleine nicht entscheidend ist, halte ich für richtig. Sonst müsste z.B. ein Geländer im Krankenhaus auch als MP klassifiziert werden. Denn wenn es zusammenbricht, ist der Schaden hoch.
Entscheidend ist also weiterhin die Frage, ob das Produkt der Diagnose, Therapie, Überwachung usw. dient.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner
…toll, Ihre Kompetenz in der Sache und ihre Bereitschaft, bei „quälenden“ Fragen vieler Menschen zu helfen.
Meine Frage: Wenn ich in meiner Dokumentations- Befundungssoftware Laborwerte aus dem KIS des Krankenhauses lade und ohne Empfehlung/Deutung zur Verfügung stelle, z.B. bevor eine ANGIO gemacht wird, macht das meine Software zum MP? Im Grunde genommen könnte ja ein falscher Wert durch HL7 Übermittlungsfehler z.B. Folgen haben. Oder soll ich diese Funktion besser rausnehmen.
Danke für Ihre Hilfe
Dr. Achim Kredteck
Danke für die netten Zeilen, lieber Dr. Kredteck!
Laut MEDDEV 2.1/6 macht eine reine Anzeige von Daten eine Software noch nicht zum Medizinprodukt.
Die Frage, ob etwas passieren kann, ist bei der Klassifizierung unerheblich.
Falls die Software mehr macht als Daten unverändert anzuzeigen, abzuspeichern, abzufragen oder anzuzeigen, entscheidet nicht die Funktionalität, sondern die Zweckbestimmung.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
als KIS Hersteller ist insbesondere der letzte genannte Punkt für uns von großer Relevanz.
In erster Linie wird ein KIS ja auch lediglich zur Dokumentation genutzt,
dann wäre es auch laut MDR weiterhin kein Medizinprodukt, soweit richtig? Aber wenn bestimmte Module zur Therapie gedacht sind, fielen diese dann unter das MDR, also z.B. eine Therapieplanung oder eine Pflegeplanung. Und nur diese Teile wären dann als Medizinprodukt zuzulassen, aber nicht das ganze KIS. Liege ich damit richtig?
Mit freundlichen Grüssen
Klaus Malmede
Sehr geehrter Herr Malmede,
die MDR ändert in der Tat (fast) nicht, was die Qualifikation von Produkten als MP (oder eben nicht) betrifft.
Es ist auch zutreffend, dass Module zur Therapieplanung alleine als MP „zugelassen“ werden können, nicht aber notwendigerweise das ganze KIS.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
ein potenzielles Medizinprodukt der Klasse I, das ich in Ihren detaillierten Klassifizierungsbeispielen für Stand-Alone-Software (herzlichen Dank dafür!) ebenfalls leider nicht gefunden habe, ist die Gruppe der Übungs-/Trainings-Software, die speziell nach neurologischen Ausfällen für ein häusliches Training beeinträchtigter kognitiver Funktionen von Bedeutung ist, weil praktisch keine andere Übungsform als hochfrequentes Üben eine Aussicht auf Erfolg bietet. Konkrete Beispiele: Anklicken von Bildern nach Benennung durch Computer, schriftliche Benennungen von Bildern, Punkte auf Bildschirm suchen, …
Dass sie in die Klasse I hineingehört, dürfte unbestritten sein, aber wo beginnt die für den Klassifizierungsaufwand beim Inverkehrsbringer entscheidende Einstufung in Klasse I* im Gegensatz zur relativ einfach zu handhabenden Selbstklassifizierung ohne benannte Stelle nach I (ohne Zusatz)? Sind Ergebnisdarstellungen in Form von „High-Scores“ oder qualitative Bereichsdarstellungen, die nur der Selbstinformation des Übenden dienen, bereits eine Messfunktion?
Mit freundlichem Gruß,
Petra Roosen
Sehr geehrte Frau Roosen,
danke für Ihre Frage!
Sie fragen, wann Sie in die Klasse I* fallen würden. Meines Erachtens kommt nur ein Im in Frage. Wie man diese Klassifizierung vornimmt habe ich im MEDDEV 2.1/5: Medizinprodukte mit Messfunktion beschrieben.
Meine starke Vermutung ist, dass Sie nicht als Produkt mit Messfunktion zu klassifizieren sind.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
zunächst herzlichen Dank für die schnelle Antwort und den zielsicheren Verweis auf die spezifische „Messfunktions“-Interpretation!
Ich habe mir Ihren Starterpack heruntergeladen und versuche anhand der ausgesprochen umfangreichen, darin enthaltenen Informationen nun zusammenzupuzzeln, wie das konkrete Prozedere einer Anmeldung eines einfachen Klasse-I-Softwareprodukts denn nun vonstatten zu gehen hat. Speziell die MDR-Checkliste enthält ja extrem viele Punkte, die für Klasse-I-Software irrelevant ist.
Wenn ich es richtig verstehe, läuft eine Produktanmeldung auf folgende Schritte hinaus:
# Ein Deckblatt mit extrem kurzer Inhaltsbeschreibung, Affiliierung und Verantwortlichkeitszuweisung als (formloses?) Anmeldungsformblatt erstellen
# Eine technische Dokumentation gemäß der Übersichtsgrafik „…/Dokumentation/Technische-Dokumentation_DE.pdf“ erstellen, wobei auch hier viele der angesprochenen Punkte für Klasse-I-Software irrelevant erscheinen.
# Sich selber (auf der Basis der positiv zusammengetragenen Informationen in der technischen Dokumentation) die CE-Kennzeichnung zuweisen.
Anschließend umnebelt sich jedoch aktuell mein Verständnis der weiteren Vorgehensweise:
# Ist die obige Zusammenstellung der Notwendigkeiten vollständig?
# Was mache ich / an wen wende ich mich mit diesem Material, wenn keine benannte Stelle einzuschalten ist? Wäre das z.B. eine mit der MPG-Überwachung betraute Stelle einer Bezirksregierung?
Sollten Sie für diesen wohl vergleichsweise „harmlosen Zulassungsfall“ auch bereits eine dedizierte Infoseite erstellt haben, wäre ein kurzer Hinweis darauf auf jeden Fall sehr hilfreich!
Mit freundlichem Gruß,
Petra Roosen
Kurzer Nachtrag: Die Stelle, an der wir uns als Entwickler eintragen und das Produkt anmelden müssen (https://www.dimdi.de/dynamic/de/medizinprodukte/informationssystem/) habe ich bereits in der Zwischenzeit gefunden. Bliebe aber insbesondere noch die Frage nach dem Umfang und der Struktur der zusammenzustellenden Informationen.
Mit freundlichem Gruß,
Petra Roosen
Sehr geehrte Frau Roosen,
die Unterlagen, die die technische Dokumentation umfassen muss, haben Sie bereits gefunden.
Dann bleiben die Anforderungen an ein QM-System. Dessen Umfang variiert abhängig davon, ob Sie nach MDD oder MDR Ihr Produkt in den Verkehr bringen. Die ISO 13485 gibt Ihnen eine gute Übersicht.
Sie finden bei uns auch einen Artikel zum Thema Verfahrensanweisung für QM erstellen.
Bei einem Klasse-I-Produkt benötigen Sie keine Zertifizierung des QM-Systems.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
ich möchte ein Kommunikationshilfsmittel für Behinderte in das Hilfsmittelverzeichnis eintragen lassen.
Dieses besteht aus einem Microsoft-Tablet-Computer, einer Software als Medizinprodukt und einem Case.
Für den Microsoft-Tablet-Computer steht mir die EU-Konformitätserklärung von Microsoft zur Verfügung, welche scheinbar ausreichend ist.
Der Herr von der Zertifizierungsstelle teilte mir mit:
—
Die CE Konformitätserklärung allein für das Tablet ist in Ordnung.
Darüber hinaus müssen Sie als Hersteller des Kommunikationshilfsmittels eine CE-Konformitätserklärung zu dem Gesamtprodukt, also Hardware, Software und ggf. spezielle Cases für das Tablet erstellen und ein entsprechendes Konformitätsbewertungsverfahren durchführen.
—
Trotz umfangreicher Recherche werde ich aus der Aussage nicht schlau.
Können Sie mir sagen, welches Konformitätsbewertungsverfahren hier in Frage kommt? Muss ich dieselben Tests, die Microsoft für das Gerät durchgeführt hat, noch einmal durchführen?
Welches Konformitätsbewertungsverfahren wähle ich für Software als Medizinprodukt aus?
Über eine Antwort würde ich mich freuen.
Dankeschön.
Tom Weber
Sehr geehrter Herr Weber,
für jede EU-Richtlinie bzw. jede EU-Verordnung, die für Ihr Produkt anwendbar ist, müssen sie die jeweiligen Konformitätsbewertungsverfahren durchlaufen. Wenn Sie für Ihr Produkt die MDR bzw. MDD erfüllen müssen, weil Ihr Produkt ein Medizinprodukt ist, müssen Sie die entsprechenden Konformitätsbewertungsverfahren dieser Regularien durchlaufen. Welche das konkret sind, hängt von der Klasse des Produkt ab.
Im Starterkit, das Sie sich von der Startseite unseres Webauftritts herunterladen können, finden Sie weitere Informationen. Wenn Sie dann noch Fragen haben, nutzen Sie unser kostenloses Microconsulting, ebenfalls auf unserer Startseite.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
vielen Dank für das Erstellen dieser hilfreichen Website. Leider konnte mir der Inhalt bei folgendem Problem nur bedingt weiterhelfen:
Ich möchte mit einer Software ein Medizinprodukt der Klasse IIb steuern, ohne einen medizinischen Zweck zu verfolgen (auch die Kombination aus Beiden nicht, es geht hierbei nur um eine Fernsteuerung des MP für Übungszwecke). MPG und MDR sagen beide aus, dass es sich bei der Software in diesem Fall um kein Medizinprodukt handelt, solang kein medizinischer Zweck verfolgt wird. Ebenso Punkt 3.2 des MDCG 2019-Dokuments. Allerdings bezieht sich das Dokument unterPunkt 3.3 „Software driving or influencing the use of a medical device“ nur auf den medizinischen Zweck der „Steuerung“. Da dieser Zweck ebenfalls nicht vorhanden ist, wird unter „Note“ angegeben, dass die Software durch die MDR abgedeckt ist, obwohl mit der Steuerung kein medizinischer Zweck verfolgt wird.. Ist dies tatsächlich der Fall, obwohl selbst auch mit dem Medizinprodukt kein medizinischer Zweck verfolgt wird?
Ich freue mich über eine Antwort.
Mit freundlichen Grüßen
Markus Heimel
Sehr geehrter Herr Heimel,
meines Erachtens ist ihre software kein Medizinprodukt, sondern ein Zubehör. Allerdings bin ich nicht ganz sicher, was Sie mit „Übungszwecke“ meinen. Wenn jemand eine konkrete Behandlung übt, ist das etwas anders als wenn jemand die Bedienung eines Geräts unabhängig von einem spezifischen Patienten üben will.
Dass Zubehör keinen eigenen medizinischen Zweck verfolgt, ist der Sache inhärent.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
vielen Dank für Ihre schnelle Antwort. Als Nachtrag möchte ich hinzufügen:
Mit der Steuerung soll die Anwendung des Gerätes patientenunabhängig geübt werden.
Bezüglich Ihrer Aussage, müsste als „Zubehör“ nicht trotzdem die medizinische Zweckbestimmung des Gerätes verfolgt werden, also dass das Zubehör laut Artikel 1 Abs. 2 MDR die Verfolgung der medizinischen Zweckbestimmung des Medizinproduktes unterstützt? Wie ist es aber wenn die Steuerung dies eben nicht tut, also das MP in Kombination mit der Steuerung nur für patientenunabhängige Übungen und Ausbildungen genutzt wird und somit keinen medizinischen Zweck verfolgt?
Vielen Dank für Ihre Rückmeldung.
Mit freundlichen Grüßen
Markus Heimel
Sehr geehrter Herr Heimel,
dann ist es weder ein Medizinprodukt noch ein Zubehör. Die MDR ist somit ebenso wenig anwendbar wie die MDD.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
ein riesiger Knackpunkt der sich bei uns in der Entwicklung gerade stellt ist die Kombination einer Software (App, Klasse IIa/b)-Produkt mit der wir Puls, Schritte, Strecke messen wollen. Wir benötigen also ein(e) geeignete programmierbare(s) wearable/smartwatch für iOS und Android-Geräte. Das Produkt muss dann gemeinsam zertifiziert werden. Kann man das ohne die in den Verkehr-bringende Fa. schaffen (in dem man ein geeignetes wearable für sich auswählt) oder benötigt man auf Grund der notwendigen Zugriffe auf Quellcode/Programmierung/Data safety auf alle Fälle eine entsprechend geregelte Kooperation?
Mit freundlichen Grüßen,
Andreas Schnitzbauer
Sehr geehrter Herr Schnitzbauer,
mir fehlt möglicherweise noch etwas der Kontext, um qualifiziert antworten zu können.
Daher kann ich nicht beurteilen, ob man das System aus Software und Wearable in der Tat als ein Produkt in den Verkehr bringen muss. Das würde ich nämlich versuchen zu vermeiden. Wenn Sie eine SmartWatch als Medizinprodukt oder als Teil dessen in den Verkehr bringen, müssen Sie die Anforderungen u.a. der IEC 60601-Familie erfüllen.
Ob diese Entkopplung und der „Nicht-Zugriff“ auf den inneren Aufbau des Hardware-Produkts (inkl. dessen Software) möglich ist, entscheidet sich auch im Risikomanagement.
So weit meine ersten Gedanken ohne tieferes Verständnis des Produkts, seiner Zweckbestimmung und seiner Risiken.
Beste Grüße, Christian Johner
Vielen Dank Prof. Johner für die wertvolle Erklärung über SaMD.
Ich habe zu dem Thema eine Frage:
Gilt eine Smartwatch (z.B. Apple Watch) als Medizinprodukt. Wir haben hier 3 Komponente (Die Uhr mit den Sensoren für die Messungen und die Software, die diese Messungen interpretiert und das Handy, auf dem die App heruntergeladen wird).
1. Gilt in diesem Fall die Uhr als Medizinprodukt oder als Zubehör und wenn als Zubehör gilt, ist ein Zubehör ein Medizinprodukt und muss klassifiziert werden anhand MDR Klassifizierregeln.
2. Gilt das Handy in dem Fall auch als Zubehör?
3. Ist die Software als Standalone-software zu betrachten? (die Software interpretiert die Messungen, die von den Uhrsensoren gemessen werden)
Vielen Dank, dass Sie Ihre wertvolle Erfahrung im Bereich medizinische Zulassungen mit uns teilen.
Guten Tag Herr Muaz,
ob es sich um ein Medizinprodukt handelt oder nicht hängt von der Zweckbestimmung ab. Eine Smartwatch ist per se kein Medizinprodukt. Erst wenn sie vom Hersteller dafür bestimmt ist einen spezifischen medizinischen Zweck zu erfüllen (z.B. Diagnose, Verhütung, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten vgl. MDR Art.2 (1)) wird sie zu einem Medizinprodukt. Also z.B. wenn die Zweckbestimmung der Smartwatch mit der App lautet: „Zur Therapie und Rehabilitation bei Herzkreislauferkrankungen bei Erwachsenen Menschen zwischen 18 und 60 Jahren.“
Um ein Zubehör (vgl. MDR Art. 2 (2)) handelt es sich, wenn es die Zweckbestimmung des Medizinprodukts unmittelbar unterstützt. Ein Zubehör ist per Definition kein Medizinprodukt wird aber von der MDR so behandelt wie eines (vgl. MDR Art. 1 (1)). Das würde man beim Handy in diesem Fall aber nicht so sehen, da es auch (fast) egal ist um welches Handy es sich handelt, es stellt nur eine Umgebung dar, auf der die Software laufen kann.
Es gäbe die Smartwatch, auf der eine embedded Software läuft und die App, die man als Standalone Software betrachten könnte. Das Medizinprodukt, sofern es eines wird, könnte aber aus der Kombination von Smartwatch und App bestehen auch wenn es physisch getrennt ist.
Viele Grüße
Daniel Reinsch
Sehr geehrter Herr Prof. Johner,
in einer ambulanten Radiologie betreiben wir zur Bildachivierung das Open Source Archiv Conquest in Verbindung mit der OsiriX MD und IQView Befundsoftware.
Wir sind uns unsicher, ob vor allem das selbst betriebene open source Archiv Conquest die Anforderungen an Medizinprodukte in Deutschland erfüllt. Wir wäre für Ihre Einschätzung dankbar, ob bzw. unter welchen Rahmenbedingungen Conquest als Archiv für diagnostische Zwecke betrieben werden darf.
Besten Dank,
Dr. Norbert Reekers
Sehr geehrter Herr Dr. Reekers,
wenn Sie keine Konformitätserklärung haben bzw. bekommen, müssen Sie davon ausgehen, dass niemand die Konformität des Produkts erklärt hat. Damit darf das Produkt nicht zu den Zwecken eines Medizinprodukts eingesetzt werden.
Meines Wissens gibt oder gab es jedoch eine kommerzielle Version, für die der Hersteller die Konformität erklärt hat und die sonstigen Pflichten wie die Post-Market Surveillance erfüllt.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
besten Dank für Ihre prompte Rückmeldung.
Wir sind verunsichert, weil im Forum Datenschutzrecht auf Ihren Beitrag verwiesen wird und dort quasi als Resümee Ihre Beitrags zu Software als Med Produkt interpretiert wurde, dass im Gegensatz zum Viewer, das PACS Archiv kein Medizinprodukt sei.
https://www.forum-strahlenschutzrecht.de/apps/forum/frage/pacs-und-viewer-medizinprodukte-ja-oder-nein-oder-jein-alte-frage-neue-antworten/
Wir können das in dieser Deutlichkeit aus Ihrem Beitrag „Software als Medizinprodukt“ nicht erkennen. Hier bestätigen Sie zwar unter Punkt 10 d unter FDA-Bedingungen, „auch PACS zählen nicht mehr zu den Medizinprodukten.“. Ob dies auch für den Einsatz in Deutschland gilt, bleibt uns unklar.
Da wir ordnungsgemässe Viewer als MP mit Herstellersupport einsetzen, müsste demzufolge der Einsatz des Conquest Systems als reines Bildarchiv möglich sein, auch wenn dies als open source System ohne Support durch Hersteller/Drittanbieter mehr oder weniger in „Eigenregie“ betrieben wird.
Wie sehen Sie das?
Mit besten Grüßen
Norbert Reekers
Sehr geehrter Herr Dr. Reekers,
die Antwort hängt von der Zweckbestimmung ab.
Es gibt zum Glück eine Ausarbeitung der EU in diesem Dokument unter Punkt 8.4 Das ist genau, was Sie benötigen.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Dr. Johner,
wir(Krankenhaus) haben Software in Betrieb, die ein Medizinprodukt ist.
Wir möchten wissen, was wir aus dieser Sicht im Betrieb berücksichtigen müssen.
Zwei Aspekte sind für uns wichtig:
• Neueinführung einer Software die ein Medizinprodukt ist, was ist zu beachten ?
• Eine Software die ein Medizinprodukt ist, ist in Betrieb und wird genutzt, was ist zu beachten ?
Vielen Dank im Voraus
Sehr geehrte Frau E. Harchali,
das ist eine sehr wichtige Frage, die nicht schnell zu beantworten ist. Daher ein paar Tipps:
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Dr. Johner,
vielen lieben Dank für die schnelle Antwort
Viele liebe Grüße, H. Elharchali
Sehr geehrter Herr Prof. Dr. Johner,
übrigens wir beide haben einen gemeinsamen Freund der Thomas Erkert und er richtet Ihnen viele liebe Grüße aus :).
Viele liebe Grüße von mir auch
Hafsa Elharchali
Sehr geehrtes Johner-Team,
Ich studiere gerade den „best practice guide“ der Benannten Stellen, den Sie in Ihrem neuesten Newsletter genannt haben. Im Kapitel zu Software werden besondere Anforderungen an „class B and C“ … erwähnt.
Welche Klassifizierung ist damit gemeint? Bei Medizinprodukten kennen wir die Klassen I, IIa, IIb, III (sowie Is, Ir und Im), bei den IVDD die Klassen A bis D.
Ist das ein Beispiel für die von Ihnen im Newsletter genannten inhaltlichen Fehler des Papiers oder eine Software-spezifische Klassifizierung, oder Klassifizierung als IVDD?
Ich freue mich über eine Rückmeldung.
Mit freundlichen Grüßen,
R. Lichtenthal
Sehr geehrter Herr Lichtenthal,
die Norm IEC 62304 definiert Software-Sicherheitsklassen A, B und C.
A: keine Gefährdung des Patienten durch Fehler in der SW möglich
B: keine schwere Verletzung durch Fehler in der SW möglich
C: schwere Verletzung oder Tod durch Fehler in der SW möglich
Vielen Dank für die sehr schnelle Antwort
Dear Prof. Dr. Johner,
thanks a lot for your very detailed and complete information on the SW qualification topic.
What would be your opinion on the following case :
Considering :
– a medical device HW controlled by an embedded SW („main“ SW)
– update of the „main“ SW is done through a USB connector („manual“ update)
what about the qualification of a new SW („connectivity“ SW) which intended use is to allow „over the air“ update of the „main“ SW (e.g. through a connection to a backend server on which the update files are stored) if:
– the „connectivity“ SW is also embedded on the device HW (but segregated from the „main“ SW)
– Scenario A : if the possibility to update the „main“ SW through USB is removed
– Scenario B : if the possibility to update the „main“ SW through USB is kept
IMO, the „connectivity“ SW is „supporting“ the performance of the „main“ SW and should therefore qualify as a an accessory (and therefore be regulated), however this is kind of a borderline case and I could use an external view on this, especially as having this „connectivity“ module „deregulated“ could facilitate (read „lower the time“) its update release process considering that its connectivity feature will most-likely require frequent (>12x/year) updates to respond to security issues (e.g. coming from SOUPs monitoring).
Thanks in advance for your reply.
Regards
The intended use „over the air upload“ would not qualify the „connectivity“ SW as a medical device. I would also not consider it as an accessory to a medical device. Of course, the MDSW would not work if not able to install/update but you also do not consider the JTAG adapter and the flashing software an accessory to a medical device. It is not supporting the intended use in my opinion, it is just updating the software. The risk management of the MDSW („main“ SW) should take into consideration what happens if the update was not successful or potential IT security threats.
Sehr geehrtes Johner-Team,
Zählt eine App/Website als Medizinprodukt, wenn sie dazu dient, Bilder von Hautstellen an ein externes Unternehmen zu übermitteln? Die Bilder werden vom Patienten selbst mithilfe eines Dermatoskops aufgenommen. Anschließend werden die aufgenommenen Bilder über WIFI an die App/Website auf das Endgerät des Patienten geschickt. Von dort aus erfolgt der Versand der Bilder an das externe Unternehmen, welches anhand der Bilder eine Diagnose erstellt. Der Patient erhält diese Diagnose über die App.
Stellen sowohl das Dermatoskop als auch die App, die von uns als Hersteller entwickelt wurden, ein Medizinprodukt dar?
Vielen Dank für Ihre Hilfe!
Viele Grüße
Annalena Stolte
Liebe Frau Stolte,
die reine (verlustfreie) Übertragung von medizinischen Bildern qualifiziert ein Produkt noch nicht als Medizinprodukt (vgl. MDCG 2019-11). Die App würde in Ihrer Beschreibung keinen wesentlichen Beitrag zur medizinischen Zweckbestimmung leisten, sondern nur ein Kommunikationsmittel darstellen.
Ein Dermatoskop hat typischerweise eine medizinische Zweckbestimmung (vom Hersteller dafür gedacht pathologische Hautveränderungen besser erkennen und untersuchen zu können, vgl. MDR Art. 2 (1)) und wird als Medizinprodukt der Klasse 1 eingestuft.
Wenn Sie beides zusammen als System in Verkehr bringen, sollten Sie allerdings den Artikel 22 der MDR berücksichtigen.
Viele Grüße
Dear Johner Team,
If it exists a web-based application what recollects data (reprocessing cycle data, number of cycle, etc) from a steam sterilizer and washer disinfector devices and it displays it in a „nicer“ UI.
Additionally, this data can be printed and can be used to determine the quality of the sterilization/disinfection process, therefore, these data can be used to approve or reject a batch of the sterilzer device.
Can this be considered as a medical device? even if the shown data is exactly the same displayed in the sterilizer/washer disinfector device screen?
Lets say that the web app is used as a „physical indicator“ that proves that the temperature and pressure of the sterilizer device reaches the minimium temperature/pressure required for a correct sterilization
Dear Alexis,
The document to consider is MDCG 2019-11. E.g. you want to consult in the decision tree decision #3.
„Embellishment“ of the representation is a classical topic of borderline classification. We would have to analyze it more closely. But my first guess ist that you do not have a medical device according to the definition of MDR. Probably also not an accessory. As said, a closer analysis is need. Just reach out, if we may help.
Best regards, Christian Johner
Sehr geehrter Herr Prof. Johner,
für mich ist leider noch nicht ganz klar, wie das Verhältnis von Zweckbestimmung zu Funktion zueinander ist.
Stellt die MDCG 2019-11 in ihrem dritten Entscheidungsschritt auf die Zweckbestimmung des Produkts ab oder auf die tatsächliche Funktion? Stellt man auf die tatsächliche Funktion ab: Müsste in dem von Herrn Reinsch aufgeworfenen Beispiel nach dem dritten Entscheidungsschritt die Eigenschaft einer MDSW verneint werden, da keine Funktion besteht, die über die Dokumentation der Daten hinausgeht. Zur Prüfung der eigentlichen Zweckbestimmung im Rahmen des fünften Entscheidungsschritts würde man nicht mehr kommen.
Mit freundlichen Grüßen
Georg Keiber
Sehr geehrter Herr Keiber,
die Qualifizierung als Medizinprodukt bezieht sich auf die Zweckbestimmung. Der dritte Entscheidungsschritt dient dazu abzugrenzen, dass Verwaltungs- und Dokumentationssysteme wie z.B. KIS oder PVS nicht als Medizinprodukte zu betrachten sind. In den Beispielen D) 1. und 2. geht es über diese Funktion hinaus, weil die Produkte der Diagnosestellung dienen. Beispiel 3 unterscheidet genau zwischen reiner Dokumentation und der Überwachung/Kontrolle. Ein Dokumentationssystem, bei der die Software auch zur Überwachung von Krankheiten dient und nicht der Arzt alleine die Überwachung macht, indem er sich die Daten anschaut, müsste dementsprechend auch als Medizinprodukt qualifiziert werden.
Mit freundlichen Grüßen
Sehr geehrter Herr Prof. Johner,
vielen Dank für den informativen Artikel. Bei mir herrscht Unsicherheit bezüglich SOUP und Beispiel 2. In meinem SaMD soll neben einem Selbst-Training für die Nutzer auch per Link auf Online-Austauschgruppen hingewiesen werden. Die Zweckbestimmung bezieht sich nur auf das Selbst-Training. Die genannten Online-Gruppen sind nicht Teil der Zweckbestimmung, auf diese soll aber innerhalb der Software verlinkt werden. Handelt es sich dann hierbei um eine Software-Komponente? Oder ist es möglich in einem SaMD weiterführende Links einzubauen, wenn eine Abgrenzung (meinetwegen auch in der Zweckbestimmung) vorliegt.
Ich freue mich von Ihnen zu hören und wünsche eine schöne Woche
Liebe Grüße
Esther Diehm
Liebe Frau Diehm,
herzlichen Dank für Ihre spannende Frage. Aus meiner Sicht handelt es sich bei dem von Ihnen erwähnten Webservice (Online-Austauschgruppen) nicht um eine SOUP.
Eine SOUP wäre ein Webservice dann, wenn ich von dort Informationen anfordere, die ich innerhalb meiner SaMD benötige. Etwa eine bestimmte Berechnung, an welche ich verschiedene Parameter sende und ein Ergebnis erhalte.
Das erkenne ich in Ihrem Fall nicht. Wenn mit dem Klick auf den Link Ihr Produkt/Ihre SaMD verlassen wird, empfinde ich dies nicht als eine SOUP. Die Möglichkeit, das Produkt über den Link zu verlassen, würde ich jedenfalls in der Gebrauchsanweisung erklären.
Grüezi Herr Johner
In Abschnitt C, Punkt 8 führen Sie aus, dass „betriebsintern“ (z. B. innerhalb von Krankenhäusern) hergestellte Software die gleichen Voraussetzungen erfüllen müssen wie „extern“ hergestellte Software. In diesem Zusammenhang verweisen Sie auf das Swissmedic-Dokument MU101_30_008d_MB.
Dieses Dokument ist inzwischen veraltet und wurde durch das Merkblatt BW630_30_007 ersetzt. Darin wird unter Punkt 5.2 ausgeführt, dass in Gesundheitseinrichtungen hergestellte Medizinprodukte-Software die grundlegenden Sicherheits- und Leistungsanforderungen (GSLA) gemäss Anhang I EU-MDR resp. Anhang I EU-IVDR erfüllen muss, sofern sämtliche Voraussetzungen nach Art. 5 (5) EU-MDR bzw. Art. 5 (5) EU-IVDR erfüllt sind.
Die weiteren regulatorischen Anforderungen sind in diesen Fällen nicht zwingend anzuwenden, was eine gewisse regulatorische Erleichterung für betriebsintern hergestellte Software darstellt.
Freundliche Grüsse, Anja Teichmann
Grüezi, liebe Frau Teichmann!
Ihr Hinweis ist super! Damit aktualisiere ich direkt den Artikel und kann damit auf die Erleichterung hinweisen.
Herzlichen Dank!
Beste Grüsse, Christian Johner
Sehr geehrter Herr Prof. Johner,
vielen Dank für Ihren informativen und stets sehr hilfreichen Blog.
Zu diesem Blog-Beitrag bin ich in der Rolle als Betreiberin eines SaMDs gelangt. Als großer Krankenhausverbund betreiben wir natürlich ein Krankenhaus-Informationssystem, das durch den Hersteller als Medizinprodukt gekennzeichnet wurde. Im Zuge dessen sehen wir uns regelmäßig, teils mehrmals pro Monat, mit Informationen zu Updates und FSNs durch den Hersteller konfrontiert, die wir realistisch kaum zeitnah und direkt an 1000+ User schulen können. Gerne möchten wir ein Key User-/Train the Trainer-System verwenden. Nun stelle ich mir die Frage, ob dies im Einklang mit den geltenden Regularien steht?
Ebenfalls frage ich mich, ob es im Kontext von Vigilanz und PMS eine Grenze gibt, aber der viele Änderungsmeldungen oder auch FSNs „zu viele“ werden? Beim Vertrieb von Hardware als Medizinprodukten wäre eine solche Flut von Änderungsmeldungen kaum vorstellbar.
Vielen Dank im Voraus und mit freundlichen Grüßen
Anna Broich
Danke für Ihre Frage, Frau Broich!
Die Regularien geben keine spezifischen Vorgaben, wie Sie das Training realisieren, außer dass das nach den Vorgaben des Herstellers erfolgen soll. Somit gibt es weder ein Gebot noch ein Verbot des Ansatzes, das Training über die Key-User zu skalieren.
Ich würde bei der Entscheidung, wann (also wie zeitnah und wie häufig) und wie (Methodik, Didaktik) Sie das Wissen in Ihrer Organisation weitergeben, berücksichtigen:
Unabhängig davon würde ich das Thema bei den Herstellern ansprechen. Die wollen natürlich auf der „sicheren Seite“ sein und veröffentlichen daher lieber zu viel und zu häufig als zu wenig und zu langsam. Aber genau da braucht es ein Balance. Dazu können Sie beitragen.
Beste Grüße, Christian Johner
Hallo Herr Prof. Johner,
vielen Dank für die sehr schnelle und informative Antwort. Damit haben Sie unser Bauchgefühl und unsere Handlungssicherheit erhöht.
Viele Grüße
Anna Broich