Die nachgelagerte Phase: Nicht nur relevant für die ISO 14971
Eine der häufigsten Abweichungen bei Audits hat einen Bezug zur sogenannten „nachgelagerten Phase“.

Das Risikomanagement zählt zu den wichtigsten gesetzlichen Anforderungen, die Hersteller von Medizinprodukten erfüllen müssen.
Die ISO 14971 ist die Norm zur „Anwendung des Risikomanagements auf Medizinprodukte“. Sie beschreibt einen Risikomanagementprozess, der sicherstellen soll, dass die Risiken durch Medizinprodukte bekannt und beherrscht sowie im Vergleich zum Nutzen akzeptabel sind.
Abb. 1: Risikomanagementprozess nach ISO 14971 (zum Vergrößern klicken)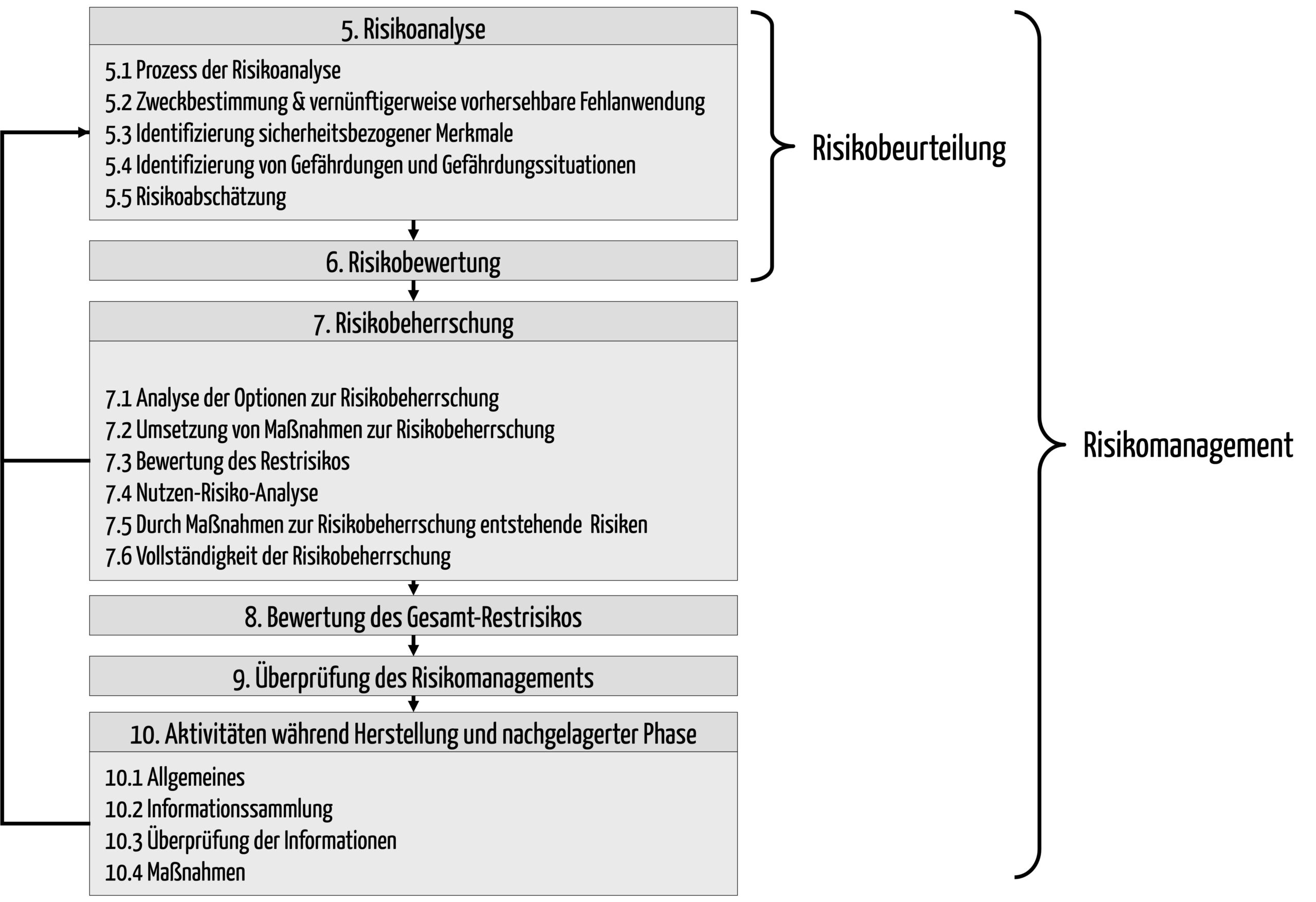
Inhalt
Sie finden auf dieser Seite Fachartikel …
Der erste Schritt beim Risikomanagement für ein konkretes Produkt besteht darin, den Risikomanagementplan zu erstellen. Dieser legt fest,
Diesen Risikomanagementplan muss die Verfahrensanweisung zum Risikomanagement einfordern
Hersteller müssen zuerst die Zweckbestimmung des Produkts festlegen.
Dann müssen sie im Rahmen einer Gefährdungsanalyse bzw. Risikoanalyse die Gefährdungen und Gefährdungssituationen identifizieren. Dazu gibt es mehrere Methoden:
Um Gefährdungen zu identifizieren, die von fehlerhaften Prozessen ausgehen, empfiehlt sich die Prozess-FMEA (pFMEA).
Im nächsten Schritt müssen die Hersteller die Risiken abschätzen. Dazu müssen sie bestimmen:
Viele Hersteller arbeiten mit einer Risikoprioritätszahl (RPZ). Dieses Konzept entspricht allerdings nicht der ISO 14971.
Spätestens jetzt besteht die Aufgabe der Hersteller darin, ihre Kriterien für die Risikoakzeptanz zu bestimmen. Die Festlegung erfolgt meist in Form einer Risikoakzeptanzmatrix.
Dabei hilft auch der Benefit-Risk-Guidance der FDA.
Bei der Bewertung des Restrisikos sollten Sie diese Zahlen kennen. Auch die Kaplan-Meier-Kurve hilft im Risikomanagement.
Gesetze wie die MDR und die IVDR verpflichten die Hersteller zur Risikominimierung bzw. Risikobeherrschung. D.h. sie müssen die Risiken so weit wie möglich und gemäß ihrer Akzeptanzkriterien reduzieren.
Sehr hilfreich bei Diskussionen mit Auditoren und Behörden sind der Artikel zu Informationen als Maßnahmen zur Risikominimierung sowie die Safety Assurance Cases, welche die FDA bei einigen Produkten sogar verlangt.
Die Ergebnisse aller bisherigen Aktivitäten müssen die Hersteller in einem Risikomanagementbericht dokumentieren. Bei dieser Bewertung sollten die Hersteller prüfen, dass sie die 7 häufigsten Fehler im Risikomanagement vermeiden.
All diese Aufzeichnungen und Dokumente bilden dann die Risikomanagementakte.
Damit endet das Risikomanagement nicht. Vielmehr folgt die Post-Market-Phase bzw. die nachgelagerte Phase, wie die ISO 14971 sie nennt. Ein Teil derer ist die Post-Market Surveillance und Überwachung der Produkte im Markt.
Die DIN EN ISO 14971 liegt in der Version 2022 vor. Die letzte wesentliche Änderung stammt aus dem Jahr 2019. Diese Änderungen wurden mit der dritten Ausgabe der ISO 14971 eingeführt. Damit wird die EN ISO 14971:2012 mit dem Annex ZA obsolet.
Im Beitrag zu den harmonisierten Normen erfahren Sie mehr zur Bedeutung der Präfixe wie DIN, EN und ISO.
Wie Hersteller die (neue Version) der Norm anwenden sollten, beschreibt der Beitrag zur ISO 24971.
Wichtig ist auch die Frage, ob die ISO 14971 überhaupt anwendbar ist. In diesem Kontext sind die Begriffe vorhersehbarer Missbrauch und anormaler Gebrauch wichtig.
In anderen Rechtsbereichen wird die ISO 31000 genutzt, die für die Medizinproduktehersteller zumindest als Anregung dienen kann.
Bei Medizinprodukten, die Software enthalten oder Software sind, wirkt sich die IT-Sicherheit wesentlich auf die Risiken aus. Hilfreich sind hier folgende Publikationen:
Für Medizinprodukte, die Software enthalten oder eine Standalone-Software sind, empfehlen sich diese Fachartikel:
Das Risikomanagement bei Medizinprodukteherstellern unterscheidet sich in manchen Aspekten von dem Risikomanagement bei anderen Organisationen:
Nutzen Sie die Unterstützung des Johner Instituts:
Melden Sie sich gleich, damit wir die nächsten Schritte besprechen können. So stellen Sie sicher, dass die „Zulassung“ sicher gelingt und Ihre Produkte schnell in den Markt kommen.
Eine der häufigsten Abweichungen bei Audits hat einen Bezug zur sogenannten „nachgelagerten Phase“.

Die HAZOP (Hazard and Operability) ist ein Gefährdungsanalyseverfahren, das die ISO 14971 neben der FMEA, FTA und PHA empfiehlt. Das deutsche Akronym für HAZOP (Hazard and Operability) lautet PAAG. Das steht für Prognose, Auffinden der Ursache, Abschätzen der Auswirkungen, Gegenmaßnahmen. Die Norm IEC 61882 beschreibt dieses Verfahren. Inhaltsübersicht HAZOP im Überblick » Vorgehen nach IEC…
DetailsWährend das Englische die Begriffe Safety und Security unterscheidet, kennen wir im Deutschen nur die Sicherheit. Zwar haben beide Begriffe mit einander zu tun, sind aber nicht deckungsgleich.

Der UL 2900-2-1 nennt sich „Particular Requirements for Network Connectable Components of Healthcare and Wellness Systems“. Er zählt zur UL-2900-Familie, der Normenfamilie zur IT-Security. Lesen Sie in diesem Artikel, welche Schwächen der Standard hat und unter welchen Umständen er Ihnen dennoch nützlich sein kann.
DetailsDer TIR 57 ist ein „Technical Information Report“ der amerikanischen AAMI. Er möchte Hilfestellung dabei geben, Risiken durch mangelnde IT-Sicherheit von Medizinprodukten zu erkennen und zu beherrschen und so die Anforderungen der ISO 14971 an das Risikomanagement zu erfüllen.
Details
Die Begriffe ‚vernünftigerweise vorhersehbarer Missbrauch‘ (‚reasonable foreseeable misuse‘), ‚anormaler Gebrauch‘ (‚abnormal use‘), ‚korrekter Gebrauch‘ (‚correct use‘) und ‚bestimmungsgemäßer Gebrauch‘ (’normal use‘) stammen aus den Normen ISO 14971 und IEC 62366-1. Leider erscheinen die Definitionen nicht gut aufeinander abgestimmt zu sein. Allerdings sind sie substantiell für das Verständnis, wie die Normen, in denen diese Begriffe erwähnt werden,…
DetailsDie ISO/IEC 15408-1 trägt den Titel „Information technology — Security techniques — Evaluation criteria for IT security — Part 1: Introduction and general model“. Sie beschreibt ganz allgemein, wie man bei der Bewertung der IT-Sicherheit z.B. von Produkten vorgehen soll. Die konkreten Hinweise, wie geprüft werden soll, finden sich in dem zweiten und dritten Teil…
DetailsAnormaler Gebrauch: Eine von vielen möglichen Fehlhandlungen Irrtum, Benutzungsfehler, anormaler Gebrauch, Aufmerksamkeitsfehler, vernünftigerweise vorhersehbarer Erinnerungsfehler, vorhersehbarer Missbrauch. Die Liste möglicher (Fehl-) Handlungen ist lang, die die Normen betrachtet haben möchten. Doch Vorsicht: Die Normen zur Gebrauchstauglichkeit (IEC 62366:2007 und IEC 62366-1:2015) und die Norm zum Risikomanagement (ISO 14971) verwenden unterschiedliche Begriffe. Dieser Artikel bringt Licht ins…
Details
Die ISO 24971 ist die Norm, die meine Auditoren-Kollegen oft zücken, wenn es mit Medizinprodukte-Hersteller Diskussionen über die Auslegung der ISO 14971 gibt. Sie kennen die ISO 24971 nicht? Sie möchten diese Norm nicht kaufen? Kein Problem, ich habe diese Technical Report für Sie gelesen und zusammengefasst.
Details
CIRS sind seit Jahren im Gesundheitswesen und der Luftfahrt gesetzlich vorgeschrieben, um Meldungen über kritische Vorkommnisse anonym sammeln, auswerten und daraus Schlüsse für ein sichereres Arbeiten ziehen zu können. Bei Medizinprodukteherstellern sind CIRS keine Pflicht. Ein Fehler? Lesen Sie, wie Sie als Medizinproduktehersteller von CIRS profitieren können.
Sie sehen gerade einen Platzhalterinhalt von Hubspot Embedded Content. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von HubSpot. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Hubspot Meetings. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr Informationen