1. Definitionen
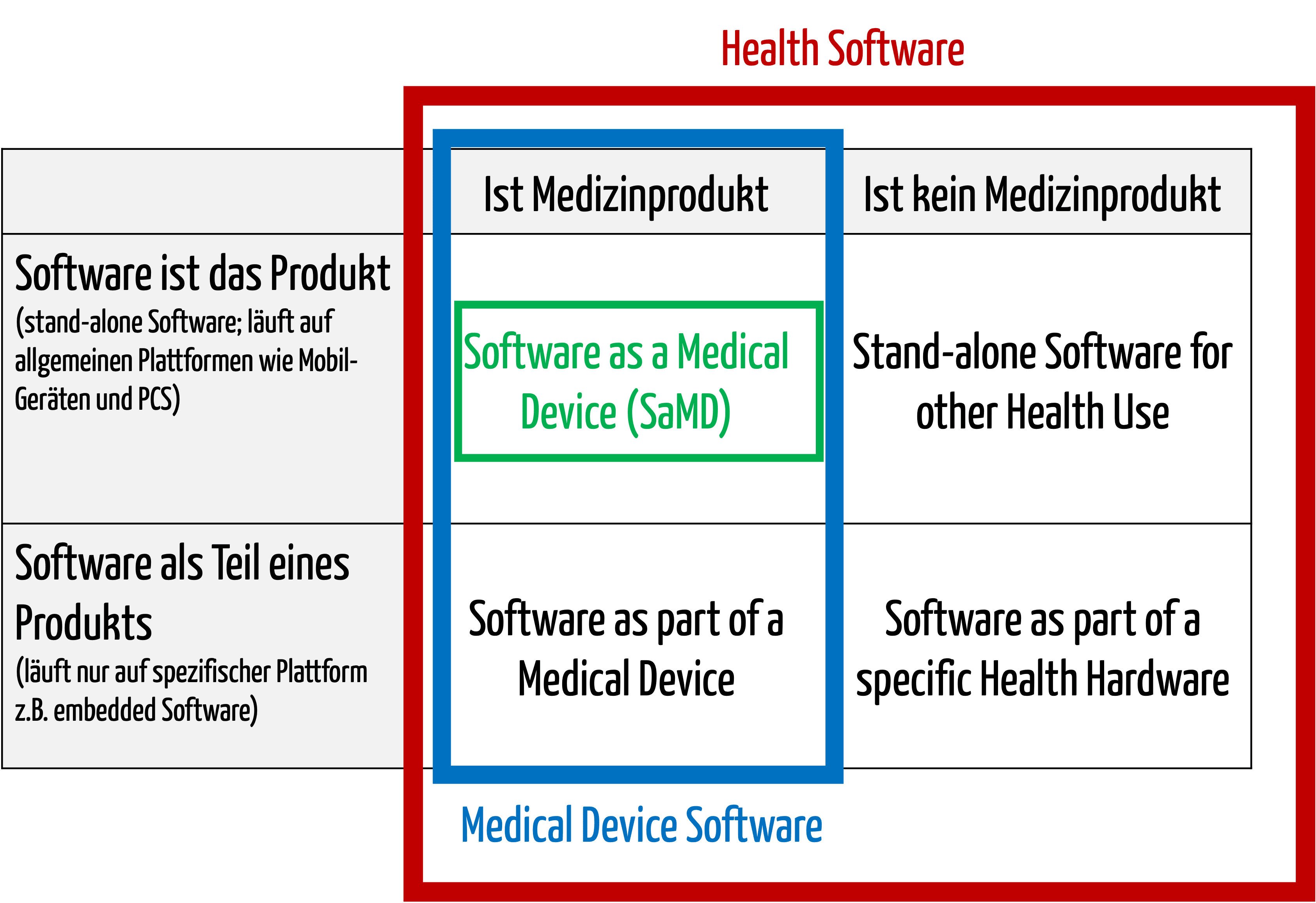
Zur medizinischen Software zählt alle Software, die für das Gesundheitswesen eingesetzt wird, insbesondere Software für Medizinprodukte bzw. Medizingeräte (Embedded Software) und Software, die selbst ein Medizinprodukt ist (Standalone-Software).
Die IEC/CD1 82304-1 (Health Software – Part 1: General requirements for product safety) unterscheidet folgende Begriffe:
- HEALTH SOFTWARE
Software intended to be used specifically for maintaining or improving health of individual persons, or the delivery of care
- MEDICAL SOFTWARE
Software intended to be used specifically for incorporation into a physical medical device or intended to be a SOFTWARE MEDICAL DEVICE
- SOFTWARE MEDICAL DEVICE
Software intended to be a medical device in its own right
- MEDICAL DEVICE SOFTWARE
Software intended to be used specifically for incorporation into a physical medical device
Damit wird klar, dass medizinische Software ein Medizinprodukt sein kann, aber nicht muss.
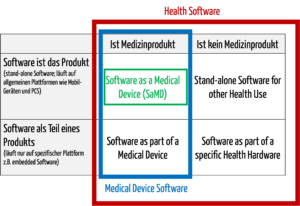
Abb. 1: Medizinische Software umfasst auch Medical Device Software und Software as a Medical Device (zum Vergrößern klicken).
Digital Health bzw. E-Health meint mehr als nur die Produkte, hier stehen die medizinischen Anwendungen und Geschäftsmodelle im Fokus.
2. Regulatorische Anforderungen
a) Medizinische Software – ein Medizinprodukt?
Es stellt sich oft die Frage, wann medizinische Software für die Medizintechnik der Definition des Begriffs Medizinprodukt entspricht. Eine weiterführende Diskussion dazu finden Sie im Artikel zur Klassifizierung von Software als Medizinprodukt sowie im Artikel zur Qualifizierung und Klassifizierung von IVD-Software.
b) Verordnungen, Gesetze, Normen
Software, die ein Medizinprodukt oder ein Teil dessen ist, muss die regulatorischen Anforderungen erfüllen:
- In Europa sind Medizinprodukteverordnungen (MDR, IVDR) relevant. Diese enthalten jedoch nur relativ allgemeine Vorschriften für Software, die dieser Fachartikel vorstellt.
- Die IEC 62304 definiert die Lebenszyklusprozesse für Software von Medizinprodukten.
- Die IEC 82304-1 ist bei jeder „Health Software“ anwendbar. Die IEC 82304-1 fordert auch Konformität mit den Anforderungen der IEC 62304.
- Es gibt zudem MDCG-Leitlinien z.B. die MDCG 2019-11 und die MDCG 2023-4.
- Die FDA stellt in ihren Guidance-Dokumenten spezifische Anforderungen, explizit auch an medizinische Software. Zudem beantwortete sie viele Fragen speziell zu Software as a Medical Device in diesem FAQ.
3. Unterstützung für Medizinproduktehersteller
Nutzen Sie die Unterstützung des Johner Instituts:
- Haben Sie Fragen zur Entwicklung und Zulassung von Medizinprodukte, die Software enthalten oder Software sind? Dann nutzen Sie das kostenfreie Micro-Consulting.
- Im Kompaktseminar Medizinische Software erwerben Sie die vorgeschriebenen Kompetenzen. Sie lernen die gesetzlichen Anforderungen an die Entwicklung von Software für die Medizintechnik kennen und erfüllen.
- Die Videotrainings des Auditgarant helfen Ihnen, Schritt für Schritt eine schlanke und IEC-62304-konforme „Software-Akte“ zu erstellen. Zusätzlich nimmt Ihnen ein vollständiger Satz an Templates viel Arbeit ab.
- Nutzen Sie auch die Unterstützung unserer Expertinnen und Experten. Sie helfen Ihnen, Ihre Software kurz, präzise und gesetzeskonform zu dokumentieren, und bereiten Sie auf Audits und „Tech File Reviews“ vor.
- Lassen Sie die IT-Sicherheit Ihrer Software durch Penetration Tests überprüfen.
Melden Sie sich gleich, damit wir die nächsten Schritte besprechen können. So stellen Sie sicher, dass die „Zulassung“ sicher gelingt und Ihre Software bzw. Ihre Produkte schnell in den Markt kommen.
Die Qualifizierung und die Klassifizierung von IVD-Software bestimmen, wie und wie schnell IVD-Hersteller ihre Software in den Markt bringen können und welche Kosten für die „Zulassung“ anfallen. Dieser Artikel unterstützt Sie dabei, IVD-Software korrekt zu qualifizieren und zu klassifizieren. Damit können Sie regulatorische Probleme sowie daraus resultierende Aufwände und Verzögerungen vermeiden.
Details
Gesetze und Normen verpflichten Medizinproduktehersteller zur Software Bill of Materials, der SBOM. Standardisierte SBOM-Formate reichen allerdings nicht immer aus, um diese Anforderungen zu erfüllen. Insbesondere Medizinproduktehersteller, die für ihre Software keine SBOM ausliefern und nutzen, sind im Markt nicht mehr akzeptiert. Hier sind die Gründe.
Details
Die Medizinprodukteverordnung (MDR) (wie bereits die Medizinprodukterichtlinie (MDD) und damit das Medizinproduktegesetz zuvor) verlangt, dass Hersteller für ihre Software Lebenszyklus-Prozesse einhalten. Auch die IEC 62304 und die IEC 82304 sprechen von Software-Lebenszyklus-Prozessen. Doch was versteht man unter einem Software-Lebenszyklus? Der Software-Lebenszyklus beinhaltet alle Phasen, die ein Software-Produkt von der ersten Idee bis zur Außerbetriebnahme durchläuft.…
Details
Unter Software-Wartung versteht man die Phase, in der die Software weiterentwickelt wird, z. B. mit dem Ziel 79% aller Bugs werden laut FDA während der Software-Wartung eingeführt. Entsprechend adressieren einige Regularien dieses Thema. Update: Ergänzung zur Software-Wartung während der Entwicklung
Details
1. Documentation Level: Ende des Level of Concern Am 14.06.2023 hat die FDA das Guidance-Dokument Content of Premarket Submissions for Device Software Functions final freigegeben. Dieses Dokument löst das Guidance-Dokument ab, welches den Level of Concern einführte und unterscheidet nur noch zwei Klassen. a) Bestimmung der Klassen Die FDA definiert nicht mehr drei „Level of…
Details
Dass Gesetze und Normen die IT-Security auch bei „Legacy Devices“ einfordern, ist verständlich. Die Art, wie diese Anforderungen formuliert werden, führt allerdings oft zu Verwirrung. Beispielsweise konnten sich Gesetzgeber und Normenkomitees nicht auf gemeinsame Definitionen einigen. So geht es einmal um die IT-Sicherheit bei Legacy Devices, einmal um die IT-Sicherheit von Altprodukten bzw. von Bestandsprodukten…
Details
Die MDCG hat im Oktober 2023 eine Leitlinie MDCG 2023-4 veröffentlicht mit dem Titel „Medical Device Software (MDSW) – Hardware combinations – Guidance on MDSW intended to work in combination with hardware or hardware components”.
Sowohl die FDA als auch die IEC 62304 kennen durch Dritte entwickelte Software. Sie sprechen von Off-the-Shelf Software (OTS) bzw. von Software of Unknown Provenance (SOUP). Worin unterscheiden sich OTS und SOUP? Welche Gemeinsamkeiten haben beide? Welche gesetzlichen Anforderungen müssen sie erfüllen? Dieser Artikel gibt Antworten.
Details
Für Hersteller ist die Antwort auf die Frage relevant, ob und wann beim Einsatz künstlicher Intelligenz in Medizinprodukten klinische Studien notwendig sind. Denn davon hängen die Dauer und die Kosten ab, um diese Produkte in den Markt zu bringen. Die gute Nachricht vorweg: Es gibt Fälle, in denen die Hersteller auf klinische Studien bei Produkten…
Details
Der Begriff „Medizinprodukte-PC“ ist nicht eindeutig definiert. Allerdings verstehen unter einem Medizinprodukte-PC die meisten Abhängig von den Konstellationen müssen die Hersteller unterschiedliche regulatorische Anforderungen erfüllen. Diese werden im vorliegenden Artikel vorgestellt.
Details