Ein Audit des Qualitätsmanagementsystems durch eine Benannte Stelle ist in den meisten Fällen die Voraussetzung dafür, dass Hersteller von Medizinprodukten ihre Produkte in Europa vermarkten dürfen.
Die FDA prüft ebenfalls Qualitätsmanagementsysteme, spricht aber nicht von Audits, sondern von Inspektionen.
Inhalt
Sie finden auf dieser Seite Fachartikel zu:
- Grundlagen zu Audits
- Ablauf von Audits
- Unterstützung bei Audits und dem Aufbau von QM-Systemen
1. Grundlagen zu Audits
a) Begriffsdefinition
Die ISO 9000:2015 definiert den Begriff Audit wie folgt:
systematischer, unabhängiger und dokumentierter Prozess zum Erlangen von objektiven Nachweisen und zu deren objektiver Auswertung, um zu bestimmen, inwieweit Auditkriterien erfüllt sind.
b) Die verschiedenen Audit-Typen
Je nach Schwerpunkt eines Audits spricht man von:
- Systemaudits (z. B. Prüfung auf Konformität eines Qualitätsmanagementsystems mit den Forderungen einer Norm wie ISO 13485 oder ISO 9001)
- Prozessaudits
- Produktaudits
- Software-Audits
Weiterhin unterscheidet man:
Beachten Sie!
Für Medizinproduktehersteller sind die Systemaudits / Audits des QM-Systems durch eine Benannte Stelle am wichtigsten. Audits durch Zertifizierstellen, die keine Benannte Stelle sind, genügen nicht, um die Anforderungen der MDR und IVDR in deren Anhängen IX nachzuweisen.
Im Erfolgsfall erhalten die Hersteller ein Zertifikat, das sie zur Inverkehrbringung ihrer Produkte in Europa berechtigt.
Beachten Sie auch den Übersichtsartikel zu den QM-Systemen, die FAQ zu QM-Systemen und Zertifizierungen sowie den Artikel zu den Benannten Stellen.
c) Regulatorische Grundlage für Audits
Europa
Ein Audit durch eine Benannte Stelle ist die Voraussetzung dafür, dass die Benannte Stelle ein Zertifikat (gemäß ISO 13485 bzw. Anhang IX) ausstellen darf. Dieses Zertifikat wiederum ist die Voraussetzung für die Inverkehrbringung von Produkten.
Vorsicht!
Hersteller dürfen nur auf Basis von Zertifikaten einer Benannten Stelle ihre Produkte in den Verkehr bringen. Es gibt weitere Zertifizierstellen, deren ISO-13485-Zertifikate dafür wertlos sind. Zudem gibt es Anbieter, die nicht einmal akkreditiert sind für die ISO 13485.
USA
Die FDA fordert inzwischen auch Konformität mit der ISO 13485. Sie verlangt aber keine Zertifikate. Sie überprüft die Konformität im Rahmen von Inspektionen. Inspektionen enden jedoch im Gegensatz zu Audits im Erfolgsfall nicht mit einem Zertifikat.
Weltweit („MDSAP-Länder“)
Das International Medical Device Regulators Forum (IMDRF) hat das Medical Device Single Audit Program MDSAP ins Leben gerufen: Mit einem MDSAP-Audit können die regulatorischen Anforderungen an die Auditierung und Inspektionen von QM-Systemen erfüllt werden.
2. Ablauf von Audits
a) Allgemeines
Die ISO 19011 beschreibt die Anforderungen an Audits (d. h. deren Planung, Durchführung und Dokumentation) sowie an die Auditoren.
Weiterführende Informationen
Weitere Vorschriften bestimmen die Dauer von Audits.
Wenn ein Auditor eine „Non-Conformity“ bescheinigt, kann das dazu führen, dass die Benannte Stelle das Zertifikat verwehrt oder entzieht. Dabei ist das, was der Auditor prüft, prinzipiell bekannt (s. Abb. 1):
- Auditoren prüfen, ob das QM-System mit seinen Vorgabedokumenten alle Aspekte der Norm abdeckt, beispielsweise ob es eine normenkonforme Verfahrensanweisung für interne Audits gibt.
- Auditoren prüfen, ob sich die Organisation an die Vorgaben des eigenen QM-Systems gehalten hat.
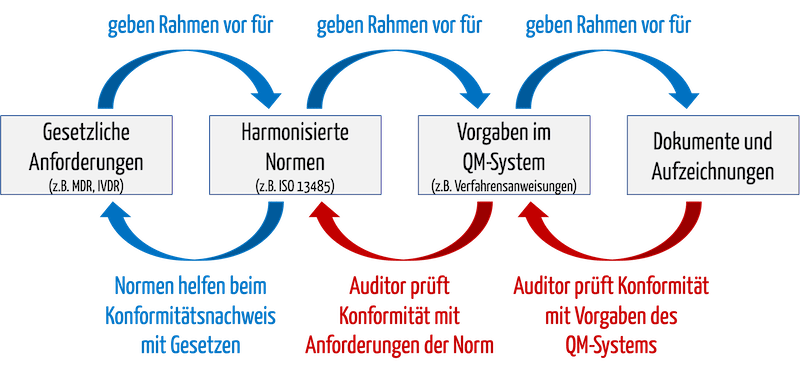
Abb. 1: Auditoren prüfen beim Audit die Konformität des QM-Systems mit normativen und gesetzlichen Vorgaben sowie die Konformität mit dem eigenen QM-System.
b) Sonderfall Offsite-Audits
In einer Bekanntmachung hat die EU genauer geregelt, wann und wie Benannte Stellen Remote-Audits durchführen dürfen:
Calls for the possibility to take temporary extraordinary measures, including remote audits, related to notified body on-site audits under the medical devices Regulations have been made by industry as well as notified bodies.
Die MDCG hat einen Guidance on temporary extraordinary measures related to medical device Notified Body audits during COVID-19 quarantine orders and travel restrictions veröffentlicht.
Darin erlaubt sie Erleichterungen, z. B.:
- Verzögerte Überwachungsaudits vor Ort
- Ersatz von Audits durch Remote-Audits
- Offsite-Inspektion / Review der Technischen Dokumentation
3. Unterstützung bei Audits
Haben Sie noch Fragen, beispielsweise zum Aufbau Ihres QM-Systems? Dann nutzen Sie unser kostenfreies Micro-Consulting.
Die Beraterinnen und Berater des Johner Instituts unterstützen nicht nur beim Erstellen von QM-Systemen, sondern prüfen diese Systeme auch und bereiten Sie auf Audits vor (z. B. mit Mock-Audits).
Interesse? Dann nehmen Sie gleich Kontakt mit uns auf.
Mit den Seminaren erwerben Sie die notwendigen Kompetenzen, um als interner oder externer Auditor zu agieren:



