Insbesondere universitäre Einrichtungen veröffentlichen medizinische Software regelmäßig als Open Source. Dabei entstehen Zweifel, ob diese Open-Source-Software als Medizinprodukt zählt und welche regulatorischen Risiken und (Produkt-)Haftungsrisiken drohen.
Dieser Artikel verschafft einen schnellen Überblick.
- Die rechtlichen und finanziellen Risiken sind davon abhängig, ob die Open-Source-Software als Medizinprodukt zählt und ob eine Inverkehrbringung vorliegt.
- Die regulatorischen Anforderungen hängen nicht davon ab, ob eine Software als Open Source in den Verkehr gebracht wird.
- Entwickler von Open-Source-Software sollten und können eine unabsichtliche Inverkehrbringung eines Medizinprodukts und die damit einhergehenden Haftungsrisiken vermeiden.
- Auch bei „Open-Source-Medizinprodukten“ muss nicht nur das Produkt, sondern auch der Hersteller/Inverkehrbringer viele rechtliche Pflichten erfüllen.
1. Rolle der Open-Source-Software in Medizinprodukten
Open-Source-Software kann sowohl das Medizinprodukt sein als auch ein Teil dessen. Möglich sind folgende Konstellationen:
- Die Open-Source-Software ist eine speziell für das Medizinprodukt entwickelte Komponente (beispielsweise ein KI-Modell mit zugehöriger API).
- Die Open-Source-Software ist eine im Medizinprodukt verwendete Off-the-shelf-Komponente wie eine Bibliothek.
- Die Open-Source-Software ist das Medizinprodukt selbst.
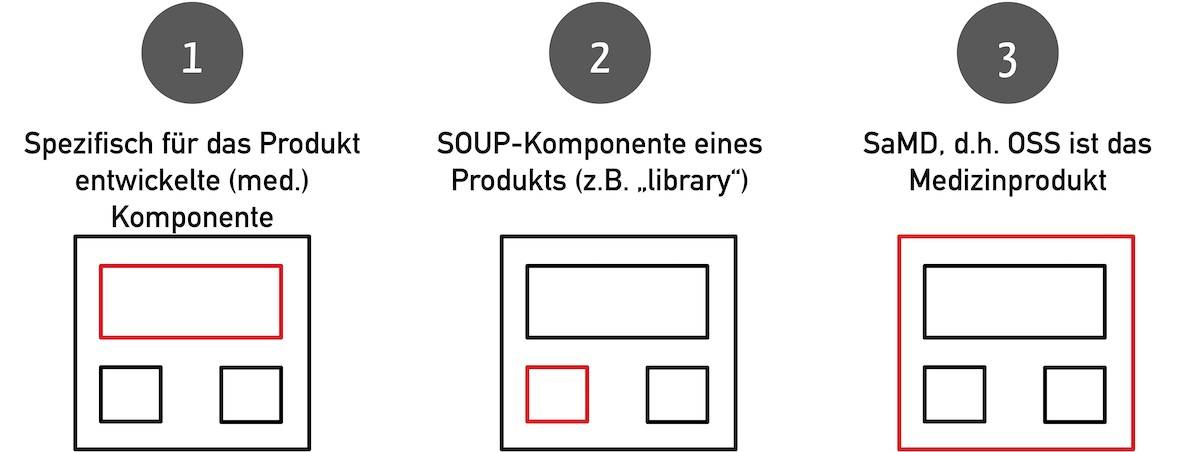
Bitte beachten Sie die Unterschiede und Gemeinsamkeiten von Off-the-shelf-Software (OTSS) und SOUP.
2. Rechtliche Einordnung
Eine Haftung entsteht insbesondere dann, wenn ein Medizinprodukt in den Verkehr gebracht wird. Daher müssen zwei Fragen geklärt werden:
- Wann zählt ein Produkt als Medizinprodukt?
- Wann liegt eine Inverkehrbringung vor?
a. Wann zählt ein Produkt als Medizinprodukt?
Nicht die Funktionalität eines Produkts entscheidet über dessen Qualifizierung (Entscheidung, ob Medizinprodukt oder nicht), sondern dessen Zweckbestimmung.
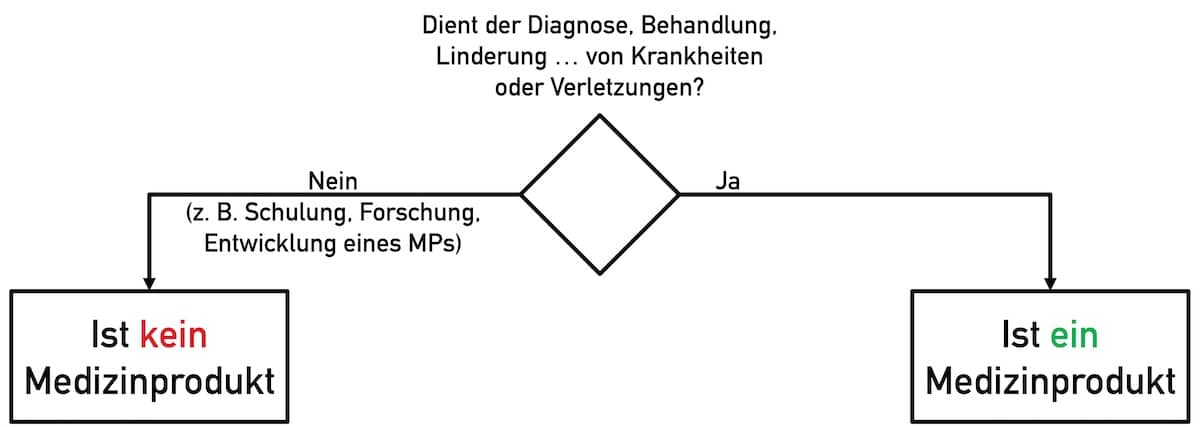
Folglich vermeiden Open-Source-Software-Entwicklerinnen beispielsweise bei den folgenden Zweckbestimmungen, dass ihre Arbeitsergebnisse als Medizinprodukt zählen:
- Das Produkt dient der Forschung bzw. der Dokumentation von Forschungsergebnissen.
- Die Software soll/darf genutzt werden, um damit Medizinprodukte zu entwickeln.
- Die Open-Source-Software dient der Ausbildung.
Besonders hilfreich ist der Hinweis, dass die Software kein Medizinprodukt sei und nicht unbesehen der Diagnose, Linderung, Therapie oder Vorhersage von Krankheiten und Verletzungen diene.
Eine (bedauerliche) Ausnahme von der Regel, dass die Zweckbestimmung und nicht die Funktionalität über die Qualifizierung entscheidet, findet sich in der MDCG-Leitlinie 2019-11. Demnach zählt Software, die nur der (unveränderten) Weiterleitung, Abspeicherung und Anzeige von Daten dient, nicht als Medizinprodukt.
Die Definition dessen, was ein Medizinprodukt ist, unterscheidet sich in den verschiedenen Rechtsbereichen. Beispielsweise fällt gemäß US FD&C viele Software nicht unter das „Enforcement“ der FDA.
b. Wann liegt eine Inverkehrbringung vor?
Die MDR legt im Artikel 2 (Abschnitte 27 und 28) fest, was eine Inverkehrbringung ist:
jede erstmalige, entgeltliche oder unentgeltliche Abgabe eines Produkts […] zum Vertrieb, zum Verbrauch oder zur Verwendung auf dem Unionsmarkt im Rahmen einer gewerblichen Tätigkeit;
Artikel 2 MDR, Abschnitte 27 und 28
Der Verbrauch bzw. die Verwendung eines Medizinprodukts in einer Arztpraxis oder in einem Krankenhaus zählt als gewerbliche Tätigkeit, auch wenn diese Tätigkeit nicht gewinnorientiert ist.
Es spielt gemäß der Definition keine Rolle, ob die Abgabe entgeltlich oder – wie häufig bei Open-Source-Software – unentgeltlich ist.
Ist die Open-Source-Software nur für den Einsatz innerhalb der Gesundheitseinrichtung bestimmt, in der sie entwickelt wurde, kommt eine Eigenherstellung gemäß MDR/IVDR Artikel 5, Abschnitt 5 in Betracht.
3. Gesetzliche Pflichten
a. Medizinprodukterecht
Zählt die Open-Source-Software als Medizinprodukt, das in den Verkehr gebracht wird oder werden soll, müssen deren Hersteller alle regulatorischen Anforderungen an Medizinprodukte erfüllen.
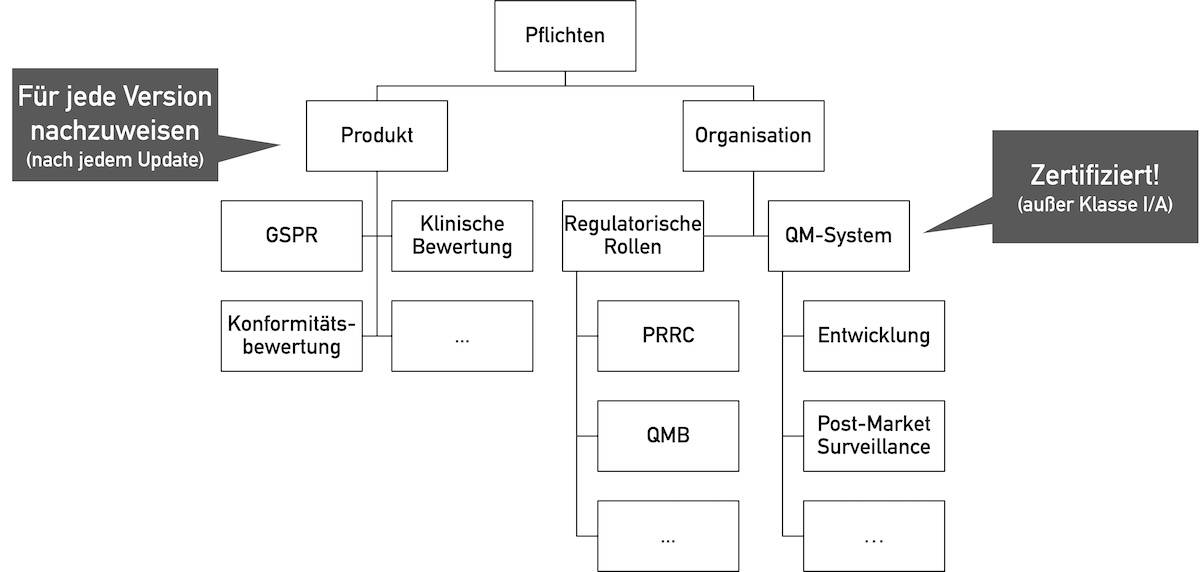
Diese Anforderungen betreffen sowohl das Medizinprodukt als auch die Organisation, welche das Medizinprodukt in den Verkehr bringt.
Die relevantesten grundlegenden Sicherheits- und Leistungsanforderungen (GSPR) an Software betreffen:
- Risikomanagement (nach ISO 14971)
- Software-Entwicklungsprozesse (nach IEC 62304)
- Usability Engineering (nach IEC 62366-1)
- IT Security (nach IEC 81001-5-1)
Zudem bedarf es eines Qualitätsmanagementsystems nach ISO 13485 bzw. Anhang IX der MDR/IVDR. Dazu zählt auch ein Post-Market-Surveillance-System.
Bedauerlicherweise bestehen viele deutsche Behörden unabhängig von den gesetzlichen Vorgaben im Anhang VIII der MDR (v. a. Regel 11) auf einer Einteilung in die Klasse IIa oder höher.
Allerdings sind die meisten gesetzlichen Pflichten wie die GSPR weitgehend unabhängig von der Klasse der Software.
Dieser Artikel beschreibt die Schritte, um ein Medizinprodukt in den Verkehr zu bringen.
b. Weitere gesetzliche Vorgaben
Zusätzlich zum und unabhängig vom Medizinprodukterecht müssen die Hersteller ggf. weitere „horizontale“ Regulierungen beachten, beispielsweise:
- AI Act
- EU Data Act
- EU Health Data Space
- IT-Security-Vorgaben wie NDIS2
- Produkthaftungsgesetz/-verordnung/-direktive
Die Anwendbarkeit und die relevanten Vorgaben dieser Vorschriften hängen von der Rolle ab, die die Open-Source-Entwickler einnehmen.
Der AI Act stellt keine Anforderungen an Open-Source-KI-Systeme.
4. Bewertung und Anwendungsbeispiele
a. Vorteile von medizinischer Open-Source-Software
Die Entwicklerinnen und Entwickler von Open-Source-Medizinprodukten argumentieren mit folgenden Vorteilen:
- Open Source fördert die Innovation und bringt Innovation schnell bis ans Patientenbett.
- Die Entwicklungsergebnisse stehen allen zur Verfügung, unabhängig von deren finanziellen Möglichkeiten.
- Die Transparenz und der Community-Ansatz helfen, Fehler schnell zu finden und zu beseitigen, was der Patientensicherheit dient.
- Es gibt keinen Interessenskonflikt wie etwa bei herstellerfinanzierten klinischen Prüfungen.
b. Herausforderungen bei Open-Source-Medizinprodukten
Die Entwicklung von Open-Source-Medizinprodukten muss spezifische Herausforderungen bewältigen:
- Es bedarf einer Organisation mit (zertifiziertem) Qualitätsmanagementsystem und allen regulatorischen Rollen wie dem QMB und der PRRC.
- Die Kompetenzen müssen auch außerhalb der Entwicklung den regulatorischen Anforderungen genügen, beispielsweise im Qualitäts- und Risikomanagement, beim Usability Engineering und in Reglatory Affairs.
- Die hohen Post-Market-Aufwände müssen über Jahre erbracht und finanziert werden, zumindest solange noch ein Produkt auf dem Markt ist.
- Da es keine Verkäufer-Käufer-Beziehung gibt, ist die Kommunikation mit den Kunden/Anwendern schwieriger, was u. a. das Risikomanagement und die Post-Market Surveillance herausfordernder gestaltet.
- Personalfluktuation, beispielsweise bei Doktorandinnen und Doktoranden, macht es herausfordernd, die Kompetenzen dauerhaft zu sichern.
- Die Lenkung (Erstellung, Prüfung, Freigabe) von Artefakten wie Dokumenten und Quellcode bedarf bei einer Community, die nicht einer Organisation entspricht, eines besonderen Augenmerks.
Das Diabetes-Dosierungssystem der Firma Tidepool basiert auf einer Open-Source-Software. Ihre regulatorische Reise hat die Firma hier beschrieben.
5. FAQ
a. Wie vermeide ich, dass ich als Open-Source-Entwickler bzw. ‑Entwicklerin verklagt werde?
Das Risiko, verklagt zu werden, kann nicht ausgeschlossen werden, aber die Wahrscheinlichkeit reduziert. Das Risiko ist umso größer,
- je offensichtlicher ein möglicher Gesetzesverstoß ist,
- je höher die ökonomische Relevanz des Produkts ist (z. B. für Mitbewerber) und
- je finanziell potenter die beklagte Partei.
Es ist deshalb hilfreich, sehr konsistent und transparent die Zweckbestimmung und Claims zu formulieren und die Anwendbarkeit regulatorischer Anforderungen zu recherchieren und zu erfüllen.
Das Johner Institut unterstützt auch Entwicklerinnen und Entwickler von Open-Source-Produkten bei der regulatorischen Strategie. Zum einen mit dem Ziel, eine Qualifizierung als Medizinprodukt entweder zu vermeiden oder diese zu erreichen, zum anderen mit dem Ziel, regulatorische Sicherheit und Konformität zu erzielen.
Melden Sie sich bei Interesse einfach über das Kontaktformular.
b. Muss ich ein CE-Zeichen anbringen? Falls ja, wo?
Mit der CE-Kennzeichnung erklärt der Inverkehrbringer die Konformität mit anwendbaren EU-Richtlinien bzw. Verordnungen. Diese Kennzeichnung ist nur gestattet und gleichzeitig gefordert, falls das Produkt in den Anwendungsbereich einer dieser Richtlinien oder Verordnungen fällt und dort die CE-Kennzeichnung gefordert wird.
Beachten Sie auch den Fachartikel mit Details zur CE-Kennzeichnung und der Anbringung des CE-Zeichens bei Software.
c. Worauf muss ich beim Einsatz von GitHub achten?
IT-Systeme für das Versions- und Konfigurationsmanagement sowie für das Betreiben von CI/CD-Pipelines zählen als computerisierte Systeme im Rahmen des QM-Systems und müssen daher einer Computerized Systems Validation unterzogen werden.
Diese Systeme müssen die gesetzlich und normativ geforderte Dokumentenlenkung sicherstellen, was jedoch eher eine Frage der korrekten Konfiguration ist und weniger der Features dieser Systeme.
d. Wie dokumentiert man ein Open-Source-SaMD für eine FDA-Zulassung?
Für Open-Source-SaMD gelten die gleichen regulatorischen Anforderungen wie für alle anderen SaMD. Dazu zählen:
- Technische Dokumentation vergleichbar mit MDR
- Cybersecurity-File als Teil der Technischen Dokumentation
- Qualitätsmanagement nach 21 CFR part 820
- Freigabe der FDA, beispielsweise im Rahmen einer 510(k) oder De-Novo-Zulassung.
e. Wer kann mir helfen?
Die (Software) Experts des Johner Instituts helfen bei allen Fragen rund um Dokumentation, Zulassung und Etablierung eines Managementsystems. Ebenso bei den vorgeschriebenen Prüfungen, z. B. Usability-Tests, Pentests und klinischen Prüfungen.
6. Fazit und Zusammenfassung
Viele universitäre Einrichtungen unterschätzen den Aufwand für eine gesetzeskonforme Entwicklung, Inverkehrbringung und Überwachung von Medizinprodukten im Markt. Ein professionelles Software-Engineering ist eine notwendige, aber bei weitem keine hinreichende Voraussetzung.
Es kann hilfreich sein, Forschung und Entwicklung zu trennen und sich einer Kommerzialisierung nicht grundsätzlich zu verschließen, zumal diese den Open-Source-Gedanken nicht ausschließt. Denn Gutes für Patienten zu tun, kostet nicht nur Zeit, sondern leider auch Geld.