
Wenn Sie ein Medizinprodukt auf den Markt bringen möchten, stellt sich schnell die Frage, wie Sie vorgehen sollten und welche gesetzlichen Regelungen Sie beachten müssen.
Dieser Artikel gibt Ihnen Antworten und stellt Ihnen die sieben Schritte vor, mit denen Ihnen die Inverkehrbringung Ihrer Produkte schnell und gesetzeskonform gelingt.
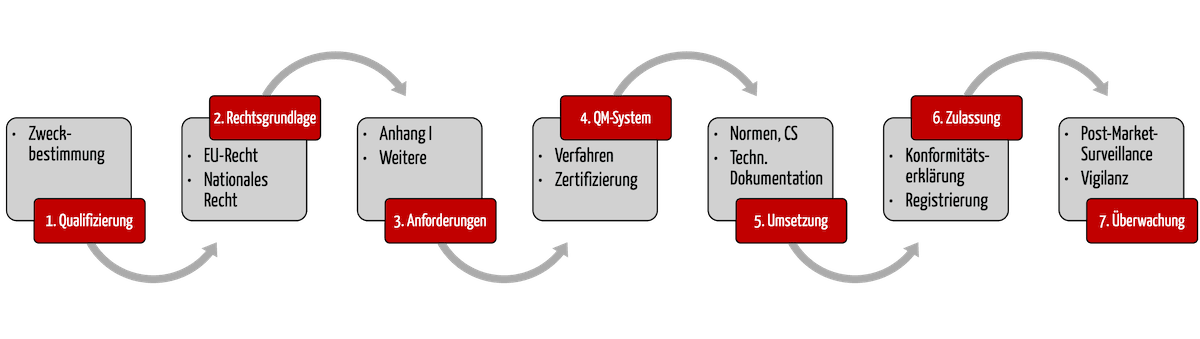

1. Schritt: Feststellen, ob das Produkt ein Medizinprodukt bzw. IVD ist
Die erste Frage, die Sie sich stellen sollten, lautet: Ist Ihr Produkt überhaupt ein Medizinprodukt? Diese Entscheidung nennt man die Qualifizierung.
Falls das Produkt kein Medizinprodukt ist, gelten dafür andere oder sogar gar keine Bestimmungen.
Entscheidend bei der Frage “Medizinprodukt ja oder nein” ist die Zweckbestimmung.
Die Zweckbestimmung des Produkts legt der Hersteller selbst fest. Er bestimmt dabei, wofür sein Produkt genutzt werden soll. Wofür man das Produkt ansonsten noch nutzen könnte oder welche Eigenschaften es mit sich bringt, ist dabei unerheblich.
Lesen Sie in diesem Artikel über Zweckbestimmung und bestimmungsgemäßen Gebrauch, was Sie dabei alles festlegen müssen.
a) Medizinprodukt nach Definition der MDR
Wird das Produkt laut Definition der Verordnung 2017/745 über Medizinprodukte (MDR) für medizinische Zwecke verwendet, ist es ein Medizinprodukt. Die MDR definiert, was ein Medizinprodukt ist:
„Medizinprodukt“ bezeichnet ein Instrument, einen Apparat, ein Gerät, eine Software, ein Implantat, ein Reagenz, ein Material oder einen anderen Gegenstand, das dem Hersteller zufolge für Menschen bestimmt ist und allein oder in Kombination einen oder mehrere der folgenden spezifischen medizinischen Zwecke erfüllen soll:
- Diagnose, Verhütung, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten,
- Diagnose, Überwachung, Behandlung, Linderung von oder Kompensierung von Verletzungen oder Behinderungen,
- Untersuchung, Ersatz oder Veränderung der Anatomie oder eines physiologischen oder pathologischen Vorgangs oder Zustands,
- Gewinnung von Informationen durch die In-vitro-Untersuchung von aus dem menschlichen Körper − auch aus Organ-, Blut- und Gewebespenden − stammenden Proben und dessen bestimmungsgemäße Hauptwirkung im oder am menschlichen Körper weder durch pharmakologische oder immunologische Mittel noch metabolisch erreicht wird, dessen Wirkungsweise aber durch solche Mittel unterstützt werden kann.
Die folgenden Produkte gelten ebenfalls als Medizinprodukte:
- Produkte zur Empfängnisverhütung oder -förderung,
- Produkte, die speziell für die Reinigung, Desinfektion oder Sterilisation der in Artikel 1 Absatz 4 genannten Produkte und der in Absatz 1 dieses Spiegelstrichs genannten Produkte bestimmt sind.”
Quelle: MDR Artikel 2
Bei der Nutzung zu Fitness-Zwecken (etwa bei einer Smartwatch) ist die Auswertesoftware kein Medizinprodukt.
Ist die Zweckbestimmung jedoch, die Daten der Software zur Diagnose oder Überwachung einer Erkrankung heranzuziehen, ist die auswertende Software ein Medizinprodukt.
b) In-vitro-Diagnostikum gemäß IVDR
Das Produkt könnte außerdem auch ein In-vitro-Diagnostikum (IVD) sein. Auch IVD sind Medizinprodukte und fallen unter die entsprechenden rechtlichen Regelungen.
Die IVD-Eigenschaft ergibt sich ebenfalls aus der Zweckbestimmung, wie die Definition aus Art. 2 IVDR verdeutlicht:
„‚In-vitro-Diagnostikum‘ bezeichnet ein Medizinprodukt, das als Reagenz, Reagenzprodukt, Kalibrator, Kontrollmaterial, Kit, Instrument, Apparat, Gerät, Software oder System − einzeln oder in Verbindung miteinander − vom Hersteller zur In-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben, einschließlich Blut- und Gewebespenden, bestimmt ist und ausschließlich oder hauptsächlich dazu dient, Informationen zu einem oder mehreren der folgenden Punkte zu liefern
- über physiologische oder pathologische Prozesse oder Zustände,
- über kongenitale körperliche oder geistige Beeinträchtigungen,
- über die Prädisposition für einen bestimmten gesundheitlichen Zustand oder eine bestimmte Krankheit,
- zur Feststellung der Unbedenklichkeit und Verträglichkeit bei den potenziellen Empfängern,
- über die voraussichtliche Wirkung einer Behandlung oder die voraussichtlichen Reaktionen darauf oder
- zur Festlegung oder Überwachung therapeutischer Maßnahmen.
Probenbehältnisse gelten als auch In-vitro-Diagnostika;“
Quelle: Artikel 2 IVDR
Die Zweckbestimmung bestimmt auch die Risikoklasse eines Medizinprodukts. Diese Risikoklasse wiederum ist u. a. ausschlaggebend dafür, welches Konformitätsbewertungsverfahren das Produkt durchlaufen muss (siehe unten).
Auf die Besonderheiten bei Zubehör und bei Software gehen wir jeweils in einem separaten Beitrag ein. Hilfreich ist auch der Beitrag zur Qualifizierung und Klassifizierung von Medizinprodukten.
2. Schritt: Rechtsgrundlage identifizieren
Ist Ihr Produkt ein Medizinprodukt, müssen Sie im nächsten Schritt herausfinden, welche rechtlichen Anforderungen das Produkt und der Hersteller erfüllen müssen. Für den deutschen Markt ergeben sich diese aus deutschem und europäischem Recht.
a) Europäisches Recht
Die Rolle der EU-Verordnungen und EU-Richtlinien
Europäisches Recht steht in der rechtlichen Hierarchie über dem deutschen Recht. Das bedeutet, dass Sie verbindliches europäisches Recht vorrangig berücksichtigen müssen.
Für Hersteller und Einzelpersonen direkt verbindlich sind EU-Verordnungen. Diese müssen Sie daher von EU-Richtlinien unterscheiden, die nicht unmittelbar für Sie gelten.
EU-Verordnungen (VO) gehören zu den unmittelbar bindenden Rechtsakten der EU. Sie müssen NICHT von den Mitgliedsstaaten umgesetzt werden, damit sie wirksam sind. Verordnungen funktionieren daher wie „europäische Gesetze“. EU-Verordnungen müssen Hersteller vorrangig zu deutschem Recht (z. B. Medizinprodukte-Durchführungsgesetz MPDG) berücksichtigen. Die Gesetze dürfen die EU-Verordnungen nur ergänzen und konkretisieren.
EU-Richtlinien: Von der EU erlassene Richtlinien (RL) sind dagegen nur für die Mitgliedsstaaten bindend. Damit sie auch für Bürgerinnen und Bürger sowie Unternehmen bindend werden, müssen sie in nationales Recht umgesetzt werden. In diesem Fall müssen sich Hersteller am deutschen Recht orientieren, das die Richtlinie umsetzt (z. B. Medizinproduktegesetz MPG), und nicht an der Richtlinie selbst.
Diese Unterscheidung ist vor allem deshalb wichtig, weil die nationalen Gesetzgeber einen Gestaltungsspielraum bei der Umsetzung einer Richtlinie haben. Viele nationale Gesetze gehen über die Anforderungen der Richtlinien hinaus.
Für Medizinprodukte relevante Vorgaben
Die wichtigsten EU-Regelungen für Hersteller sind:
- Verordnung 2017/745 über Medizinprodukte (MDR) (verbindlich)
- Verordnung 2017/746 über In-vitro-Diagnostika (IVDR) (verbindlich)
Weitere einschlägige Regelungen können sich etwa aus der Rolle als Wirtschaftsakteur und aus dem Produktportfolio ergeben:
- Verordnung 2012/207 − Verordnung über elektronische Gebrauchsanweisungen für Medizinprodukte (verbindlich, bereits teilweise aufgehoben, endgültige Aufhebung zum Mai 2024)
- Verordnung 2016/679 − Verordnung zum Schutz natürlicher Personen bei der Verarbeitung personenbezogener Daten, zum freien Datenverkehr und zur Aufhebung der Richtlinie 95/46/EG (Datenschutz-Grundverordnung) (verbindlich)
Für manche Themen existieren weiterhin EU-Richtlinien, z. B.:
- Maschinenrichtlinie RL 2006/42/EG (wenn das Medizinprodukt bewegliche Teile enthält)
- RoHS Richtlinie 2011/65 (EU) − Verordnung zur Beschränkung der Verwendung bestimmter gefährlicher Stoffe in Elektro- und Elektronikgeräten (Hier gibt es laufend Änderungen durch delegierte Richtlinien. Diese erlässt die EU-Kommission, um bestehende Regelungen zu ändern. Mehr dazu auf den Seiten der EU.)
b) Nationales Recht
Zusätzlich zum europäischen Recht müssen Hersteller nationales Recht berücksichtigen. Dieses ist vor allem relevant, wenn das nationale Recht
- komplett eigenständige Regelungen aufstellt,
- europäische Richtlinien umsetzt oder
- europäische Verordnungen ergänzt.
Auch deutsches Recht geht wie EU-Recht mit unterschiedlicher „Schärfe“ vor. Es besteht aus
- Gesetzen, z. B. dem Medizinprodukterecht-Durchführungsgesetz (MPDG),
- Verordnungen, z. B. der Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAIMV) oder der Medizinprodukte-Betreiberverordnung (MPBetreibV),
- sonstigen Vorgaben wie den technischen Richtlinien des Bundesamtes für Sicherheit der Informationstechnik (BSI).
3. Schritt: Regulatorische Anforderungen bestimmen
Haben Sie festgestellt, welche Regelungen für Ihr Produkt einschlägig sind, müssen Sie die darin gestellten Anforderungen identifizieren. In der Regel bestehen diese aus den folgenden Punkten:
a) Produkt klassifizieren
Nach der Risikoklasse Ihres Produkts richtet sich das Konformitätsbewertungsverfahren. Dies ist das Verfahren, in dem Sie nachweisen, dass Ihr Produkt die einschlägigen rechtlichen Anforderungen erfüllt.
- Die MDR unterscheidet die Risikoklassen I bis III.
- Die IVDR unterscheidet die Risikoklassen A bis D.
An Produkte mit höheren Risikoklassen stellt dieses Verfahren strengere Anforderungen als an solche mit niedrigeren.
Die Regeln für das Klassifizieren Ihres Produkts finden Sie anhand von Anhang VIII der MDR bzw. Anhang VIII der IVDR. Beachten Sie auch zugehörige Leitlinien.
Mehr über die Klassifizierung nach MDR lesen Sie im Beitrag Klassifizierung von Medizinprodukten. Weiterführendes Wissen über die Klassifizierung von IVD erhalten Sie im Beitrag Klassifizierung von In-vitro-Diagnostika: Wie Sie eine zu hohe Einstufung vermeiden.
b) Konformitätsbewertungsverfahren wählen
Abhängig von der Risikoklasse unterscheiden sich die Schritte des Konformitätsbewertungsverfahrens.
Der Artikel zu den Konformitätsbewertungsverfahren verschafft einen Überblick darüber, welche Verfahren es gibt und welche die Hersteller bei welcher Klasse durchlaufen dürfen.
c) Weitere Anforderungen bestimmen
Zu den wichtigsten Anforderungen an die Produkte zählen die grundlegenden Sicherheits- und Leistungsanforderungen (s. 5. Schritt). Weitere Anforderungen betreffen das QM-System (siehe 4. Schritt) und die Post-Market-Surveillance (siehe 7. Schritt).
Für Medizingeräte fordert die IEC 60601-1 die Bestimmung der wesentlichen Leistungsmerkmale. Eine ähnliche Anforderung stellt die ISO 14971 im Kapitel 5.3.
Beachten Sie die grundlegenden Sicherheits- und Leistungsanforderungen; der verlinkte Artikel führt in das Thema ein und enthält eine Übersicht.
4. Schritt: QM-System etablieren
Wenn Sie Medizinprodukte in Verkehr bringen, benötigen Sie ein Qualitätsmanagementsystem (QMS). Mindestanforderungen daran finden Sie in den Artikeln 10 der MDR bzw. IVDR sowie in deren Anhängen IX.
Bei Klasse I-Produkten bzw. bei IVD der Klasse A muss das QM-System nicht durch eine Benannte Stelle zertifiziert sein, in den anderen Fällen in der Regel schon:
Das bei Produkten der Klassen IIa und höher am häufigsten verwendete Konformitätsbewertungverfahren nach Anhang XI bedingt ein zertifiziertes QM-System. Andere Konformitätsbewertungsverfahren wie das nach Anhang XI Teil B sind nur in wenigen Fällen sinnvoll.
Mehr Informationen finden Sie in unserer Übersicht QM-Systeme & ISO 13485. Wie eine QM-Verfahrensanweisung aussehen sollte, erfahren Sie im Beitrag Verfahrensanweisung für QM erstellen.
Falls Sie Unterstützung dabei wünschen, das für Sie passende (schnellste, ressourcensparendste) Konformitätsbewertungsverfahren auszuwählen, dann melden Sie sich.
Sollten Sie ein ISO 13485-konformes QM-System benötigen, steht Ihnen unser Team beim Aufbau und der Einführung zur Seite.
Dabei übernehmen wir gerne auch die Rolle des externen QM-Beauftragten.
5. Schritt: Die regulatorischen Anforderungen erfüllen
Haben Sie erst einmal identifiziert, welche rechtlichen Anforderungen für Ihr Produkt und Ihre Organisation gelten, müssen Sie diese Anforderungen erfüllen (und dies auch nachweisen). Hierbei sind Normen und weitere „Beweisführungsinstrumente“ hilfreich.
a) Normen
Normen: Die Grundlagen
Mit der Hilfe von Normen können Hersteller von Medizinprodukten nachweisen, dass ihre Produkte die Anforderungen von rechtlichen Regelungen erfüllen. Normen sind Standards, die den Stand der Technik repräsentieren. Ihre Anwendung ist freiwillig. Weil sie oft weltweit anerkannt sind, erleichtern sie den Konformitätsnachweis durch die Standardisierung und die ständige Praxis ihrer Anwendung.
Normen werden von unabhängigen (nichtstaatlichen) Organisationen erarbeitet. Die Bezeichnung der einzelnen Normen weist durch ihr vorangestelltes Kürzel darauf hin, welche Organisation die Norm erarbeitet hat.
Übersicht der wichtigsten Organisationen mit Kürzel:
- DIN
- Deutsches Institut für Normung (eingetragener Verein mit Sitz in Berlin)
- EN
- Europäische Normen-Organisationen Comité Européen de Normalisation (CEN; dt.: Europäisches Komitee für Normung), Comité Européen de Normalisation Electrotechnique (CENELEC; dt.: Europäisches Komitee für elektrotechnische Normung)
- CEN: Normen zur europäischen Vereinheitlichung im technischen Bereich
- CENELEC: Normen zur europäischen Vereinheitlichung im Bereich Elektrotechnik
- ETSI
- European Telecommunications Standards Institute (ETSI; dt.: Europäisches Institut für Telekommunikation)
- Private, gemeinnützige Organisation für europäische Normen im Bereich Informations- und Kommunikationstechnologie
- ISO
- International Organization for Standardization
- Standards in allen Bereichen, die nicht durch die IEC oder die ITU abgedeckt sind
- IEC
- International Electronical Commission
- Bereich Elektrotechnik/Elektronik
- IEEE
- Institute of Electrical and Electronics Engineers
- Normen hauptsächlich zu den Bereichen Elektrotechnik und Informationstechnik
- ITU
- International Telecommunication Union
- Bereich Telekommunikation
Harmonisierte Normen
Harmonisierte Normen sind auf europäische Vorschriften abgestimmt und von den öffentlichen Stellen anerkannt. Wenn sich Hersteller an diese Normen halten, besteht daher eine Vermutungswirkung, dass hierdurch die Anforderungen von EU-Regelungen eingehalten sind.
Weitere Informationen finden Sie im Beitrag Harmonisierte Normen: Beweisführung für Medizinproduktehersteller.
Passende Normen finden
Welche Norm Sie brauchen, ist stark vom Produkt abhängig.
- Identifizieren Sie, welche Anforderungen Ihr Produkt erfüllen muss.
- Prüfen Sie, ob es für die jeweilige Anforderung eine Norm gibt. Falls es harmonisierte Normen gibt, sollten Sie diese bevorzugen.
Ein guter Startpunkt für die Recherche sind die folgenden Normen:
- ISO EN 13485: Medizinprodukte – Qualitätsmanagementsysteme – Anforderungen für regulatorische Zwecke
- ISO EN 14971: Anwendung des Risikomanagements auf Medizinprodukte
- IEC EN 62366-1: Anwendung der Gebrauchstauglichkeit auf Medizinprodukte
- IEC EN 62304: Software-Lebenszyklus-Prozesse für Medizinprodukte
- IEC EN 60601-1: Programmierbare elektrische medizinische Systeme: Basissicherheit und wesentliche Leistungsmerkmale
- DIN EN ISO 10993-1: Biologische Beurteilung von Medizinprodukten: Beurteilung und Prüfungen im Rahmen eines Risikomanagementsystems
b) Weitere „Beweisführungsinstrumente“
Common Specifications
Für einige Anforderungen von MDR und IVDR fehlen harmonisierte Normen, auf die sich die Hersteller stützen könnten. Hier kommen sogenannte “Common Specifications” (gemeinsamen Spezifikationen) ins Spiel, die die EU-Kommission festlegt und veröffentlicht. MDR und IVDR definieren die Common Specifications wie folgt:
“gemeinsame Spezifikationen” […] bezeichnet eine Reihe technischer und/oder klinischer Anforderungen, die keine Norm sind und deren Befolgung es ermöglicht, die für ein Produkt, ein Verfahren oder ein System geltenden rechtlichen Verpflichtungen einzuhalten.
Mehr Wissen hält dieser Beitrag für Sie bereit: Common Specifications – Gemeinsame Spezifikationen: Konkurrenz für die Normen?
Leitlinien und weitere Quellen
Viele Quellen sind rechtlich nicht bindend. Sie können jedoch beispielsweise bei der Interpretation von Gesetzen, Verordnungen oder Normen helfen. Hierzu gehören:
- IMDRF-Dokumente: Dokumente des International Medical Device Regulators Forum
- Manual V1.22 on borderline and classification: Manual on borderline and classification in the Community Regulatory framework for medical devices
- MDCG-Dokumente: Durchführungs- und Entscheidungshilfen für MDR und IVDR, die rechtlich nicht bindend sind. Sie werden jedoch von den Benannten Stellen in der Regel berücksichtigt. Weitere Informationen finden Sie in unserem Beitrag zur MDCG.
Zudem existieren Vorgaben, die sich auf die abgelösten EU-Medizinprodukterichtlinien beziehen, teilweise nicht mehr den Stand der Technik repräsentieren, die aber zumindest in der Übergangsphase noch relevant sind.
- MEDDEV-Dokumente: Durchführungs- und Entscheidungshilfen für MDD, die rechtlich nicht bindend sind. Weitere Informationen finden Sie in unserem Beitrag zu MEDDEV.
- NB-Med/Team NB-Dokumente: Dokumente des NB-Med (Zusammenschluss der Benannten Stellen)
2013/172/EU Empfehlung der Kommission vom 5. April 2013 über einen gemeinsamen Rahmen für ein System einmaliger Produktkennzeichnung für Medizinprodukte in der Union - ZLG-Dokumente: Antworten und Beschlüsse des Erfahrungsaustauschkreises der Benannten Stellen
- EK-Med-Dokumente: Untergruppe der ZLG
c) Grundlegende Sicherheits- und Leistungsnachweise erbringen
Nachdem Sie nun wissen, welche Normen und Regularien für Sie relevant sind, müssen Sie diese entsprechend umsetzen. Das heißt,
- entweder die gestellten Anforderungen in einen Prozess umwandeln und diesen in Ihrem Unternehmen etablieren (z. B. Software-Lebenszyklus-Prozess nach IEC 62304). Hierzu erstellen Sie Verfahrensanweisungen und zugehörige Vorgabedokumente.
- und/oder das Produkt so designen und entwickeln, dass die Anforderungen erfüllt sind (z. B. Vorgaben zu Kriechstrecken nach IEC 60601-1). Dies belegen Sie mit entsprechenden Tests.
Beispiel: Gebrauchstauglichkeit nachweisen
Als Hersteller müssen Sie die Gebrauchstauglichkeit ihres Produkts sicherstellen. Die Evaluation der Gebrauchstauglichkeit gewährleistet, dass das jeweilige Produkt von den vorgesehenen Nutzern in der vorgesehenen Nutzungsumgebung für den vorgesehenen Zweck sicher verwendet werden kann und keine nicht vertretbaren Risiken im Rahmen der Nutzung entstehen.
In den meisten Fällen ist für den objektiven Nachweis der sicheren Anwendung eine abschließende summative Studie notwendig.
Weitere Informationen dazu finden Sie im Übersichts-Beitrag Usability & IEC 62366.
d) Klinische Bewertung durchführen
In der klinischen Bewertung bzw. Leistungsbewertung bei IVD fließt alles Wissen zusammen. Sie dient der Überprüfung der Sicherheit und Leistung (einschließlich des klinischen Nutzens) des Produkts bei der vom Hersteller vorgesehenen Verwendung.
Eine Übersicht liefert der Beitrag Klinische Bewertung von Medizinprodukten.
Gegebenenfalls muss die klinische Bewertung durch eine klinische Prüfung ergänzt werden. Bei der klinischen Prüfung werden die notwendigen klinischen Daten mit dem eigenen Produkt in der überwachten Anwendung am Menschen erzeugt. Bei der Leistungsbewertung eines IVD wäre das Pendant eine klinische Leistungsstudie.
Wann eine solche klinische Prüfung nötig ist, beschreiben die Beiträge Klinische Prüfungen von Medizinprodukten: Die 7 größten Herausforderungen bzw. für IVD Leistungsbewertung von In-vitro-Diagnostika: In 8 Schritten zur Konformität.
e) UDI vergeben
Vergessen Sie nicht die Vergabe der Unique Device Identification (UDI). Durch diese Identifikationsnummer lassen sich Medizinprodukte leicht identifizieren und nachverfolgen.
Mehr zum obligatorischen UDI-System hält der Beitrag Unique Device Identification (UDI) für Sie bereit.
f) Nachweise in der technischen Dokumentation zusammenführen
Die technische Dokumentation besteht aus Dokumenten, die Hersteller von Medizinprodukten bereitstellen müssen. Sie ist die Voraussetzung für die Konformitätsbewertung und damit für die Zulassung Ihrer Medizinprodukte.
Geregelt ist die technische Dokumentation in Anhang II der MDR bzw. IVDR.
Mehr zu den Anforderungen an die technische Dokumentation finden Sie in unserer Übersicht Technische Dokumentation für Medizinprodukte.
g) Weitere Anforderungen und Nachweise
Verantwortliche Person
Sowohl die MDR als auch die IVDR fordern die Benennung einer Person Responsible for Regulatory Compliance (PRRC). Diese muss Folgendes sicherstellen:
- Die Konformität der Medizinprodukte wird in Übereinstimmung mit dem QM-System (vor deren Auslieferung) geprüft.
- Technische Dokumentation und Konformitätserklärung werden erstellt und aktuell gehalten.
- Die Post-Market Surveillance wird durchgeführt.
- Alle Meldepflichten werden erfüllt.
- Bei Prüfprodukten wird eine Erklärung gemäß Anhang XV, Kapitel 2 ausgestellt.
Die Bestellung der verantwortlichen Person ist verpflichtend. Bei Vernachlässigung dieser Pflichten drohen in Deutschland Ordnungsstrafen von bis zu 30.000 Euro.
Einzelheiten zu Aufgaben und geforderten Kompetenzen der verantwortlichen Person erfahren Sie im Beitrag MDR/IVDR – ‘Person Responsible for Regulatory Compliance’ (PRRC)”.
Insbesondere für Kleinst- und kleine Unternehmen übernehmen wir gerne die Rolle der verantwortlichen Person gemäß MDR und IVDR und sichern Ihre regulatorische Compliance.
Post-Market Surveillance Plan
Zur technischen Dokumentation zählt auch der Plan für die „Überwachung nach der Inverkehrbringung“ (Post-Market Surveillance). Mehr dazu ist in Schritt 7 beschrieben.
6. Schritt: Konformität erklären & Produkt ‚zulassen‘
Wenn Sie alle nötigen Anforderungen erfüllt haben, müssen Sie nur noch wenige Punkte berücksichtigen, bevor Sie Ihr Produkt auf den Markt bringen dürfen.
a) Konformität erklären
Mit der Konformitätserklärung erklären Sie als Hersteller, dass Ihr Produkt den rechtlichen Vorgaben entspricht.
Nachdem Sie sichergestellt haben, dass alle notwendigen Vorgaben erfüllt sind, stellen Sie eine Konformitätserklärung für Ihr Produkt aus. Hierfür benötigen Sie oft Zertifikate einer Benannten Stelle.
Was genau die Konformitätserklärung beinhaltet, erfahren Sie im Beitrag EU-Konformitätserklärung – Declaration of Conformity.
b) Als Hersteller registrieren
Hersteller müssen sich bei der Europäischen Datenbank für Medizinprodukte (EUDAMED) registrieren.
Mehr dazu erfahren Sie im Beitrag EUDAMED: European Database on Medical Devices.
c) Produkt registrieren
Darüber hinaus müssen Hersteller ihr Produkt beim Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) bzw. über die EUDAMED registrieren.
7. Schritt: Produkte im Markt überwachen
Wenn Sie Ihr Produkt auf den Markt gebracht haben, sind Sie weiterhin für dessen Sicherheit und Leistung verantwortlich. Entsprechende Prozesse müssen Sie bereits im Qualitätsmanagementsystem verankern. Hierzu gehören insbesondere:
a) Post-Market Surveillance
Auch nachdem Sie Ihr Produkt auf den Markt gebracht haben, müssen Sie es überwachen.
Die Post-Market Surveillance (Überwachung nach der Inverkehrbringung) ist ein proaktiver und systematischer Prozess, um aus Informationen über Medizinprodukte, die bereits in Verkehr gebracht wurden, notwendige Korrektur- und Vorbeugemaßnahmen (CAPA, corrective and preventive action) abzuleiten.
Mehr über Post-Market Surveillance erfahren Sie im Beitrag Post-Market Surveillance und Überwachung der Produkte im Markt.
b) Vigilanz
Vigilanz bedeutet, dass jedes „schwerwiegende Vorkommnis“ sowie jede sicherheitsrelevante Korrekturmaßnahme den zuständigen Behörden offiziell gemeldet werden muss. Im Gegensatz zur Post-Market Surveillance ist Vigilanz daher reaktiv statt proaktiv.
Die Meldepflichten sind in der MDR, Art. 87 sowie Art. 82, verankert.
Mehr zum Thema finden Sie im Beitrag Vigilanz-System.
Unsere digitalen Lösungen unterstützen Sie zuverlässig bei Ihrer Post-Market Surveillance sowie dem Regulatory Monitoring und nehmen Ihnen die manuelle Arbeit ab.
Der Regulatory Radar hält Sie über alle regulatorischen Änderungen und Neuerungen auf dem Laufenden.
Mit dem Post-Market Radar können Sie sicher sein, keine wichtigen Meldungen mehr zu Ihren eigenen sowie Vergleichsprodukten zu verpassen.
Fazit und Zusammenfassung
Um ein Medizinprodukt auf den Markt zu bringen, müssen Hersteller zahlreiche rechtliche Regelungen berücksichtigen. Auf den ersten Blick kann dies überwältigend erscheinen. Die skizzierten sieben Schritte bilden eine Richtschnur, mit deren Hilfe Sie Ihr Medizinprodukt sicher durch die Zulassung bringen können.
Sollten Sie dennoch unsicher sein oder weitere Fragen haben, können Sie sich an Ihre Benannte Stelle wenden oder das Johner Institut kontaktieren.
Änderungshistorie
- 2025-04-07: Hinweise in Kapitel 4, 5 und 7 ergänzt
- 2023-05-09: Grundlegende Überarbeitung
- 2021-10-01: Artikel initial veröffentlicht



Sehr geehrter Herr Kollege Johner,
Ich wäre Ihnen verbunden, wenn Sie nach Streichung der MPSV, also auch der in § 14 Abs. 1 S. 3 enthaltenen Pflicht zu planerischer Vorsorge für etwa nötig werdende Rückrufe eines Medizinprodukts, unterstützten: Ist eine solche Vorsorge auch heute noch ausdrücklich vorgesehen? In der MDR jedenfalls konnte ich eine Nachfolgeregelung bislang nicht finden.
Mit freundlichen Grüßen
Ulrich Foerste
Sehr geehrter Herr Kollege Foerste,
danke für Ihre spannende Frage!
Dass ein Hersteller alles dafür tut, um im Fall eines sicherheitsrelevanten Vorfalls schnell reagieren zu können, hält die MDR für wichtig. Das drückt sie im Erwägungsgrund 32 im letzten Satz aus. Dort fordert sie ein „System“ dafür. Die Forderungen nach der UDI dienen ebenfalls diesem Ziel.
Im Artikel 10 (Abschnitt 9, Aufzählungspunkte k) f.) fordert die MDR ein Verfahren für die Meldung von Sicherheitskorrekturmaßnahmen und das Management korrektiver Maßnahmen. Solche Verfahren würde ich als „Planung“ verstehen.
Da diese Maßnahmen „unverzüglich“ zu erfolgen haben (u.a. Artikel 87), wird eine entsprechende Planung eher unumgänglich sein. Ohne Planung funktionieren die Kommunikationskanäle meist nicht, da nicht klar ist, wie Anwender und Betreiber erreicht werden. Auch gibt es keine Klarheit im Unternehmen darüber, nach welcher Algorithmik die Maßnahmen ausgewählt werden.
Konnte ich Ihre Frage damit beantworten? Falls nicht, haken Sie gerne nach.
Beste Grüße, Christian Johner
Sehr geehrter Herr Kollege Johner,
besten Dank für die Wegweisung! Ich meine ebenfalls, dass diese Vorschriften schon hinreichenden Anhalt für eine Vorbereitungspflicht geben, zumal wenn man noch Art. 83 Abs. 2 MDR einbezieht.
Mit freundlichen Grüßen
Ulrich Foerste