Unter der Technischen Dokumentation (auf Englisch „Technical File“) versteht man alle Dokumente, die Hersteller von Medizinprodukten bereitstellen müssen. Die Technische Dokumentation ist die Voraussetzung für die
Konformitätsbewertung und damit für die Zulassung der Medizinprodukte.
1. Regulatorische Anforderungen an die Technische Dokumentation
a) Medizinprodukte-Richtlinie 93/42/EWG (MDD)
Die Medizinprodukte-Richtlinie (93/42/EWG) spezifiziert Anforderungen an Medizinprodukte, u. a. die sogenannten grundlegenden Anforderungen. Hersteller sind gesetzlich verpflichtet, die Einhaltung dieser Anforderungen nachzuweisen. Dieser Nachweis erfolgt in Form der Technischen Dokumentation (auch Technical Dokumentation (TD) oder Technical File genannt).
Die Medizinprodukte-Richtlinie formuliert (im Gegensatz zur MDR) die Anforderungen an die Dokumente selbst nicht so präzise. Sie zählt aber auf, was diese abdecken müssen:
- Allgemeine Beschreibung des Produkts, einschließlich der geplanten Varianten, und seiner Zweckbestimmung;
- Konstruktionsunterlagen, einschließlich der anzuwendenden Normen und der Ergebnisse der Risikoanalyse sowie einer Beschreibung der Lösungen zur Einhaltung der für die Produkte geltenden grundlegenden Anforderungen, falls die in Artikel 5 genannten Normen nicht vollständig angewendet werden;
- Techniken zur Kontrolle und Prüfung der Auslegung, der Verfahren und der systematischen Maßnahmen, die bei der Produktauslegung angewendet werden;
- bei einem Produkt, das seiner Zweckbestimmung gemäß an ein anderes Produkt angeschlossen werden muss, der Nachweis, dass das erstere Produkt bei Anschluss an ein anderes Produkt, das die vom Hersteller angegebenen Merkmale aufweist, die grundlegenden Anforderungen erfüllt;
- die gewählten Lösungen gemäß Anhang I Kapitel I Abschnitt 2;
- die präklinische Bewertung;
- die klinische Bewertung gemäß Anhang X;
- der Entwurf der Kennzeichnung und gegebenenfalls der Gebrauchsanweisung.
All diese Unterlagen müssen Hersteller im Rahmen des Konformitätsbewertungsverfahrens vorlegen – als Technische Dokumentation.
b) Medizinprodukte-Verordnung MDR
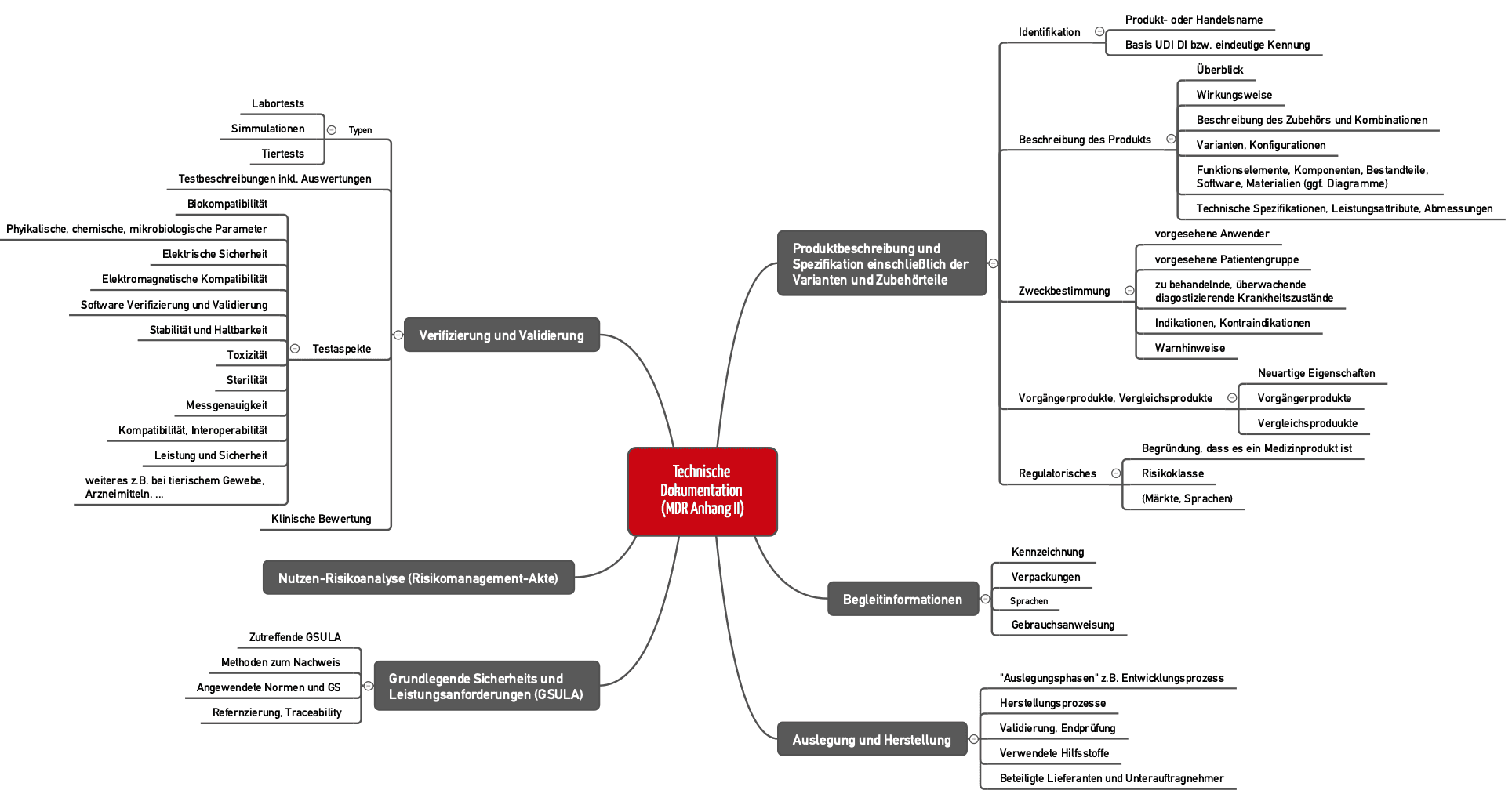
Die Medizinprodukte-Verordnung (MDR) legt nicht nur Anforderungen an das Produkt (Anhang I), sondern auch an die Dokumentation selbst fest (Anhang II). Diese muss umfassen (s. Abb. 1):
- Identifikation des Produkts (z. B. durch UDI)
- Beschreibung des Produkts inklusive dessen „wichtigsten Funktionselementen“
- Beschreibung von Produktvarianten, Konfiguration und Zubehör
- Zweckbestimmung
- Labeling (Verpackung, Gebrauchsanweisung etc.)
- Informationen zur Auslegung und Herstellung des Produkts
- Risikomanagementakte
- Verifizierung und Validierung des Produkts und damit Nachweis, dass die grundlegenden Sicherheits- und Leistungsanforderungen erfüllt sind
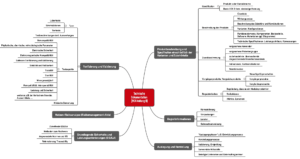
Abb. 1: Die MDR spezifiziert in Anhang II die Anforderungen an die Technische Dokumentation (zum Vergrößern klicken).
Was sind die „wichtigsten Funktionselemente“?
Mancher fragt sich, was die MDR unter den „wichtigsten Funktionselementen“ (Functional Key Elements) versteht.
Hier erwartet der Gesetzgeber (insbesondere bei den aktiven Produkten) eine grobe Systemarchitektur und damit eine Beschreibung der wichtigsten Komponenten und deren Funktionalität. Wenn relevant (z. B. weil im Kontakt mit dem menschlichen Körper), will man auch die Zusammensetzungen („Rezepturen“) der Produkte und Komponenten erkennen können.
All das entspricht dem perfekten Input für eine FMEA auf der ersten, maximal zweiten Bausteinebene. Mit dieser Beschreibung kennt man die Komponenten des Produkts (inkl. deren Funktionalität, Zusammensetzung und Interaktion) und kann damit bei der FMEA analysieren, welche Risiken entstehen, wenn sich die Komponenten nicht spezifikationsgemäß verhalten, z. B. nicht die spezifizierte Funktionalität aufweisen.
Neben der FMEA dient die Beschreibung der „wichtigsten Funktionselemente“ auch ganz banal dazu, das Produkt grob zu verstehen. Es geht dabei nicht um Geschäftsgeheimnisse, sondern darum, dass der TD-Reviewer das Gerät nicht am Tisch hat und aufschrauben kann, um es zu verstehen. Genau deshalb wird diese Beschreibung benötigt.
Die MDR geht sogar noch einen Schritt weiter: Unter der Technischen Dokumentation subsumiert sie die Marktüberwachung nach der Inverkehrbringung (Post-Market Surveillance) mit ihrer Planung und Durchführung. Anhang III legt die entsprechenden Anforderungen fest.
c) ISO 13485:2016
Die ISO 13485 hat mit der Version 2016 die Medizinprodukteakte eingeführt. Diese Akte muss ähnliche Informationen bereitstellen:
- Beschreibung des Produkts
- Zweckbestimmung
- Labeling (Verpackung, Kennzeichnung, Gebrauchs-, Installations- und Instandhaltungsanweisungen)
- Spezifikation des Produkts
- Spezifikation seiner Herstellung, Verpackung und Lagerung
- Marktüberwachung
- Konformitätsnachweise, u. a. Verifizierung und Validierung
d) Benannte Stellen
Auch die Benannten Stellen (Notified Bodies) haben Empfehlungen veröffentlicht, etwas die NB-MED/2.5.1. Diese Veröffentlichungen haben zwar keinen gesetzlichen Charakter; deren Inhalte werden in Audits und Prüfungen der technischen Dokumentation dennoch regelmäßig eingefordert.
e) FDA
Die FDA fordert ebenfalls eine ausführliche Dokumentation des Produkts. Sie unterscheidet dabei mehrere Akten:
f) Kanada
Basierend auf der STED-Struktur haben die kanadischen Behörden ihre Vorstellung von der Struktur der Technischen Dokumentation publiziert.
2. Inhalt der technischen Dokumentation
Die oben genannten Regularien legen die Aspekte fest, die eine Technische Dokumentation enthalten muss. Sie beschreiben aber nicht:
- Ganz konkrete Inhalte (Muss eine Software-Architektur ein Komponenten-Diagramm enthalten?)
- Aufteilung der Inhalte auf Dokumente
- Aufbau der Dokumente (Kapitelstruktur)
Vorgaben zu den konkreten Inhalten geben Normen wie die ISO 14971, die IEC 62304, die IEC 60601-1 und die IEC 62366-1. Beispielsweise legt die IEC 62366-1 fest, dass die Dokumentation die Planung einer formativen Bewertung enthalten muss. Solch granularen Anforderungen finden sich weder in der MDD, der MDR noch in der ISO 13485.
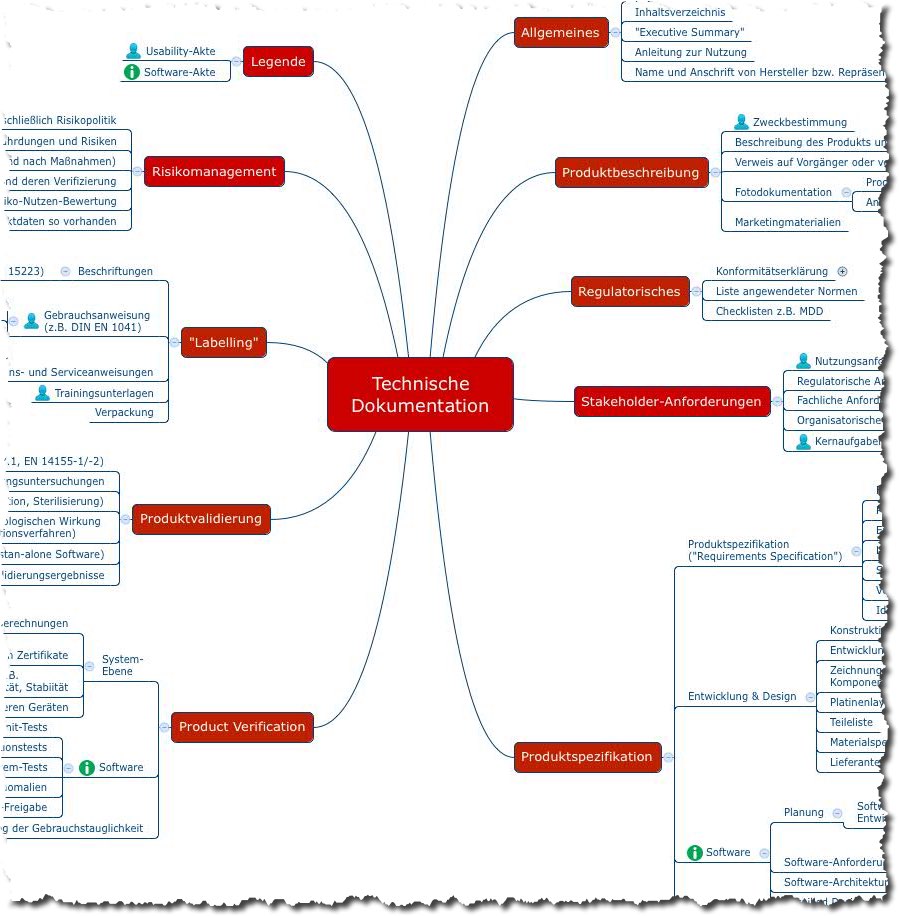
Das folgende Mindmap bietet einen Überblick über die Dokumente, die Hersteller von Medizinprodukten im Rahmen einer Konformitätsbewertung erstellen müssen. Damit weisen sie nach, dass ihre Medizinprodukte die grundlegenden Sicherheits- und Leistungsanforderungen gemäß Anhang I der Medizinprodukteverordnung (MDR) erfüllen.
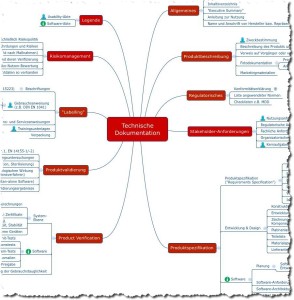
Abb. 2: Mindmap mit Inhalten der Technischen Dokumentation (Sie können sich diese Mindmap als Teil des Starter-Kits kostenlos herunterladen.)
3. Struktur der Technischen Dokumentation
Die regulatorischen Vorgaben legen nicht fest, wie Hersteller die Technische Dokumentation strukturieren müssen. Eine Ausnahme bildet die FDA, die z. B. für Premarket Notifications (510(k)) die Kapitelstruktur einschließlich Nummerierung der Kapitel vorgibt.
a) Ziele einer „standardisierten“ Struktur
Einige Player haben Vorschläge für den Aufbau einer Technischen Dokumentation erarbeitet. Sie möchten damit folgende Ziele erreichen:
- Die Hersteller erlangen schnell eine möglichst vollständige Übersicht dessen, was sie dokumentieren müssen.
- Behörden und Benannte Stellen können sich dank einer einheitlichen und logischen Struktur schnell in der Technischen Dokumentation zurechtfinden und diese prüfen.
- Hersteller haben weniger Aufwand damit, für die verschiedenen Rechtsbereiche die Akten immer neu zusammenzustellen.
b) Vorschläge für die Strukturierung der Technischen Dokumentation
STED: Summary Technical Documentation
Einer der bekanntesten Vorschläge für die Strukturierung von Technischen Dokumentationen stammt von der IMDRF (ehemalige GHTF). An dieser sogenannten STED (Summary Technical Documentation) orientieren sich viele Behörden und Benannte Stellen.
Weiterführende Informationen
Lesen Sie hier mehr zum Thema STED-Format.
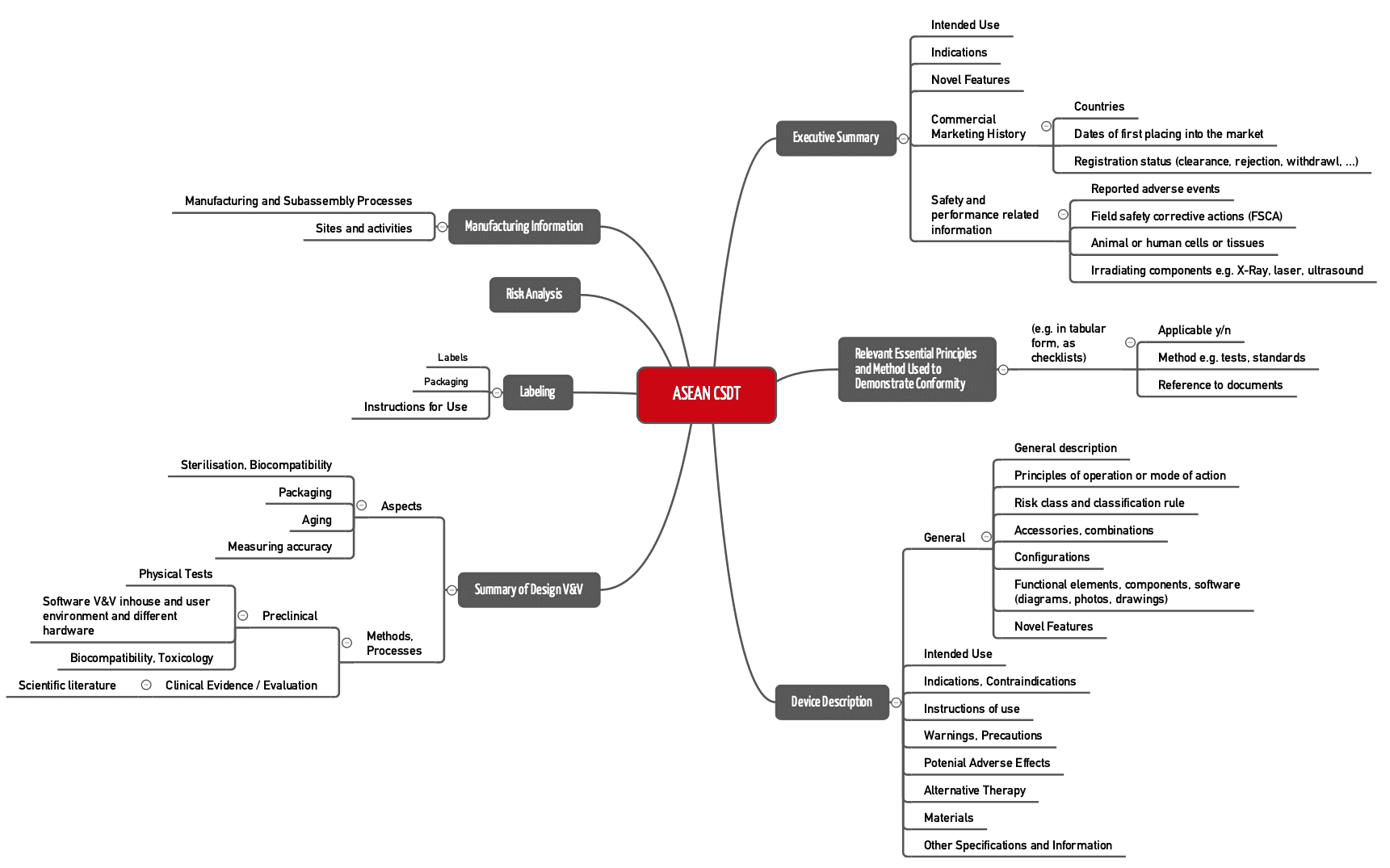
ASEAN CSDT
Die Association of Southeast Asian Nations (ASEAN) hat ebenfalls einen Vorschlag für den Aufbau von Technischen Dokumentationen erstellt, das Common Submission Dossier Template (CSDT).
Dieses ASEAN CSDT nennt sich „Guidance on Preparation of a Product Registration Submission for General Medical Devices using the ASEAN Common Submission Dossier Template”.
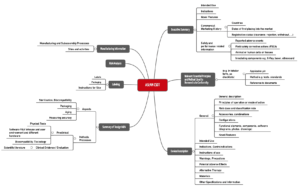
Abb. 3: Struktur einer Technischen Dokumentation nach Vorstellung des ASEAN CSDT (zum Vergrößern klicken).
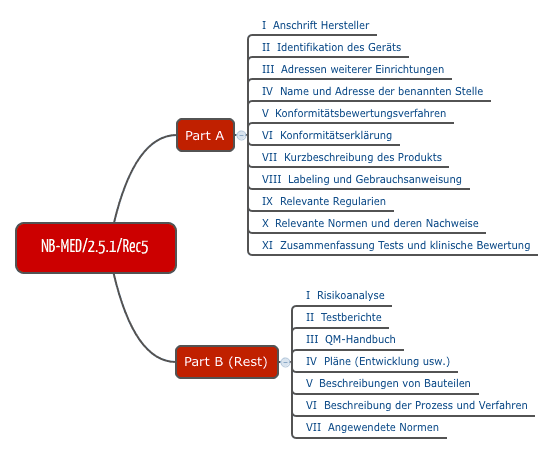
NB-MED
Ebenfalls für die Praxis relevant ist der Vorschlag des Teams NB, den man im Dokument NB-MED 2.5/1 findet.
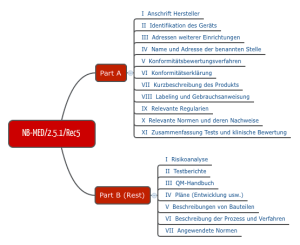
Abb. 4: Struktur der Technischen Dokumentation nach Vorstellung des Teams NB
Johner Institut
Speziell für Hersteller aktiver Medizinprodukte ist die bereits in Abb. 2 vorgestellte Struktur geeignet: Sie ist kompakt sowie verständlich und trägt maßgeblich dazu bei, die regulatorischen Anforderungen zu erfüllen.
4. Technische Dokumentation für mehrere Rechtsbereiche
Viele, aber nicht alle internationale Behörden akzeptieren die standardisierten Formate wie STED. Dennoch gibt es länderspezifische Unterschiede.
Damit Hersteller die Übersicht nicht verlieren, empfiehlt sich die Erstellung einer Mapping-Tabelle. Diese sollte alle Dokumente oder Aspekte enthalten, die im jeweiligen Rechtssystem zu adressieren sind.
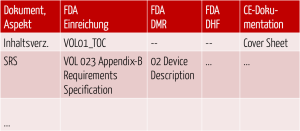
Abb. 5: Gegenüberstellung der Dokumente und Anforderungen der verschiedenen Rechtsbereiche
Die Spalten, die für die oben genannten Gliederungen stehen, beschreiben, wo (in welcher Akte) welcher Inhalt zu finden ist.
Die Aufgabe, solche Tabellen zu pflegen und die Zulassungsunterlagen spezifisch für die einzelnen Märkte zusammenzustellen, fällt der Abteilung Regulatory Affairs zu. Die Inhalte müssen hingegen die jeweiligen Fachabteilungen erstellen.
Benötigen Sie Unterstützung?
Wünschen Sie Unterstützung beim Erstellen oder Prüfen Ihrer Technischen Dokumentation? Professor Johner und sein Team helfen gerne!
Mehr erfahren
5. Zusammenspiel zwischen Technischer Dokumentation & QM-System
Die Hersteller müssen die Technische Dokumentation für ihre Medizinprodukte immer erstellen und (außer für Produkte der Klasse I) bei den Benannten Stellen einreichen. Dort wird sie erstmalig geprüft.
Die Technische Dokumentation ist auch Gegenstand der ISO 13485-Audits. Die Auditoren beurteilen anhand dieser Dokumentation,
- ob die grundlegenden Anforderungen der Medizinprodukte-Richtlinie eingehalten wurden,
- ob die Hersteller konform mit dem eigenen Qualitätsmanagementsystem arbeiten.
Schließlich muss das QM-System die Prozesse festlegen (z.B. für die Entwicklung und das Risikomanagement), mit deren Hilfe die Medizinprodukte entwickelt und produziert werden.
Vorsicht!
Die Technische Dokumentation ist nichts, was die Hersteller nachträglich für die Benannten Stellen zusammenstellen. Vielmehr ist das Technical File ein Satz an Dokumenten, die „automatisch“ und entwicklungsbegleitend geschrieben werden.
Mit anderen Worten: Ohne eine konsistente, normenkonforme und vollständige Dokumentation können Hersteller von Medizinprodukten weder den Nachweis erbringen, dass ihr Medizinprodukt die grundlegenden Anforderungen erfüllt, noch, dass ihr QM-System wirksam ist. Bei den meisten Herstellern ist das QM-System aber die Voraussetzung für die Konformitätsbewertung.
6. Unterstützung bei der Erstellung Ihrer Technischen Dokumentation
Das Johner Institut ist darauf spezialisiert, Medizinproduktehersteller beim Erstellen der Technischen Dokumentation zu unterstützen.
Haben Sie Fragen? Benötigen Sie Unterstützung? Wir helfen gerne, schnell und kosteneffizient. Erfahren Sie mehr!
Kontakt aufnehmen (via Webformular)